1 Introduction
The first recombinant adeno-associated virus (AAV) vectors were generated using both the protein capsid and the inverted terminal repeat (ITR) DNA sequence of AAV serotype 2 [1–4]. AAV2 vectors remain widely used today, and nearly all recombinant AAV genomes carry the AAV2 ITR sequence. However, a large number of alternative capsid variants have been identified from humans, baboons, chimpanzees, and rhesus, pigtailed, and cynomolgus macaques [5, 6]. These alternative capsids often have distinct tropism and antigenic profiles, although many have yet to be thoroughly studied. Traditionally, capsid variants are categorized according to their serotype. Each serotype is defined as an antigenically distinct viral capsid, as determined by serum cross- neutralization. In addition, several strategies can be used to alter the tropism of the AAV capsid, including chemical modification of the virus capsid, production of hybrid capsids, peptide insertion, capsid shuffling, directed evolution, and rational mutagenesis. The large array of capsid variants generated by these strategies, along with techniques that can enhance or alter their native tropisms, provides a rapidly expanding toolkit for gene transfer to the central nervous system (CNS). However, the number of options can be overwhelming, making it difficult to select the appropriate AAV vector for a specific application.
The recombinant AAV vectors described in this chapter can all be prepared using the same technique, described in Chapter 7, with the unique capsid sequence provided in trans during production. The basic T = 1 icosahedral architecture of the viral capsid does not differ among these serotypes and engineered vectors, although the proteins encapsidating the recombinant DNA are slightly different, resulting in limited structural changes. For many AAV serotypes cellular surface receptors or binding determinants have been identified, including sialic acid for AAVs 1, 4, 5, and 6 [7, 8], heparan sulfate proteoglycan (HSPG) for AAV2 [9], the laminin receptor for AAV8 [10], and galactose for AAV9 [11, 12]. In addition, human fibroblast growth factor receptor 1 and alphaV-beta5 integrin have both been proposed as co-receptors for AAV2 [13, 14], as has platelet-derived growth factor receptor for AAV5 [15]. These differences in receptor binding among capsid serotypes contribute to differences in tropism within the brain and other tissues. However, while differences in receptor affinity can drive variability among AAV serotypes, most, if not all, AAVs demonstrate broad tropism without absolute specificity, in part due to the wide presence of AAV receptors throughout the body. Different AAV variants can, however, differ in absolute levels of gene transfer to a specific tissue, as well as in their relative transduction strength among multiple tissues.
Several techniques can be used to generate novel AAV capsids with unique, targeted tropism. Chemical modification of the viral capsid with receptor-binding moieties can confer enhanced tropism, and chemical masking of native receptor-binding moieties can alter the normal tropism of AAV and shield the capsid from neutralizing antibodies. Hybrid capsids can be generated by co- expressing cap genes from different serotypes during production, combining the unique properties of both parental serotypes. Peptide insertion of novel receptor-binding elements on the capsid surface can alter the native tropism of AAV, and insertion of fluorescent proteins can be used to tag vector particles. Capsid shuffling and directed evolution can be used to create and screen a library of unique capsid variants for a desired trait, such as tropism for a specific cell type. Finally, rational modification of the viral capsid via site-directed mutagenesis can alter tropism, confer evasion of neutralizing antibodies, and increase transduction efficiency.
In this chapter, we describe the differing tropisms of AAV serotypes in the CNS and retina, the various factors that can influence AAV tropism, the techniques which can be used to alter the tropism of the vector, and the engineered variants that have been developed for use in the nervous system. This will provide an in-depth guide for selecting the optimal capsidserotype or engineered variant for specific experimental or therapeutic applications in the CNS.
2 Selection of the Capsid Serotype
Nervous cell tropism varies among AAVcapsid serotypes. In primary cultures of rat nervous cells, AAV5 appears to possess a strong glial tropism, and gene expression rarely colocalizes with the neuronal marker NeuN [16]. AAV serotypes 1, 2, 6, 7, 8, and 9 transduce both neurons and astrocytes in primary culture [16, 17]. AAVs 1, 6, and 7 appear to have the strongest neuronal tropism in vitro, with 75 % or more of transduced cells representing neurons [17]. AAV9, however, has relatively weak neuronal tropism in vitro, with less than 50 % of transduced cells representing neurons [17]. AAV5 is therefore recommended for transduction of cultured astrocytes, and AAVs 1, 6, and 7 are recommended for transduction of cultured neurons.
Following intraparenchymal brain injection, AAVs 1, 2, 5, 7, 8, 9, and rh.10 all exhibit strong neuronal tropism, as gene expression rarely colocalizes with markers of astrocytes or oligodendrocytes [18–21]. However, others have observed astroglial transduction with AAVs 1, 2, 5, 6, and 8 [22–24], and AAV8 has also been observed to transduce oligodendrocytes within the cortex [24]. AAV4 possesses strong glial tropism in vivo, and primarily drives gene expression within glial fibrillary acidic protein (GFAP)-positive astrocytes [25]. In addition, AAVrh.43 appears to possess stronger glial tropism in vivo than AAV8 [26]. Thus, while most AAV serotypes exhibit strong neuronal tropism following direct intraparenchymal brain injection, glial transduction has been observed in some cases, and AAVs 4 and rh.43 appear to possess stronger astroglial tropism than most AAV serotypes.
While most AAV serotypes appear to preferentially transduce neurons within the brain, the relative strength of neuronal transduction varies greatly. When compared against other serotypes, AAVs 2 and 4 typically mediate weaker and less widespread neuronal gene expression [19, 22, 27–31]. Thus, AAVs 2 and 4 are not recommended for widespread transduction of neurons. AAV2 diffuses less readily through both the brain and spinal cord parenchyma when compared against other serotypes, and therefore mediates transduction over a smaller area [19, 28, 32, 33]. This property can be harnessed for the targeting of small nuclei. The strongest and most widespread neuronal transduction is observed with AAV serotypes 1, 9, and rh.10 [19, 21, 23, 30, 34, 35]. Several novel serotypes, including pi.2, rh.8, hu.11, hu.32, and hu.37, also appear to mediate strong and widespread neuronal transduction in the brain [36]. However, these serotypes have not yet been extensively tested, and can also transduce glia within the white matter [36]. AAV serotypes 1, 9, and rh.10 are therefore recommended for targeting of neurons via intraparenchymal brain injection. Furthermore, AAV2 is recommended for the targeting of small brain regions due to its reduced diffusion in brain tissue.
Tropism also varies among AAV serotypes when administered to the cerebrospinal fluid (CSF), either via intrathecal or intra-cerebroventricular injection. AAV4 strongly transduces ependy-mal cells when administered to the ventricles, and demonstrates greater ependymal cell tropism than AAVs 2 or 5 [22]. Intracerebroventricular injection of AAV4 is therefore recommended for targeting of ependymal cells. In contrast, AAV7 and AAV9 can bypass the ependymal layer following intrathecal injection, penetrating the parenchyma and transducing neurons throughout the cortex, cerebellum, and spinal cord [37, 38]. AAVs 1, 2, 4, 5, 6, and 8 do not appear to extensively penetrate the brain parenchyma when administered to the CSF [22, 39, 40]. However, AAV6 mediates widespread transduction of spinal motor neurons following intrathecal injection [40]. Further, AAV8 possesses a unique tropism for large-diameter neurons of the dorsal root ganglion (DRG), and drives specific gene expression within these cells following intrathecal injection [39]. Thus, CSF injection of AAV4 can mediate transduction of epithelial cells, CSF injection of AAV7 or 9 can mediate widespread transduction of cortical, cerebellar, and spinal neurons, CSF injection of AAV6 can mediate transduction of spinal motor neurons, and CSF injection of AAV8 can mediate transduction of large-diameter DRG neurons.
When the spinal cord is targeted directly via intraparenchymal injection, AAVs 1, 5, and 9 demonstrate the strongest neuronal tropism, while AAVs 2, 6, and 8 demonstrate weaker neuronal tropism [19, 40]. AAV8 retains its tropism for large-diameter DRG neurons after intraparenchymal injection [39]. AAVs 1, 5, and 9 are therefore recommended for intraparenchymal targeting of discrete populations of spinal motor neurons, while AAV8 is recommended for intraparenchymal targeting of large-diameter DRG neurons.
When administered intravenously, AAVs 9, rh.10, rh.8, and rh.43 can penetrate the blood–brain barrier and drive gene expression throughout the nervous system [41–44]. These serotypes possess both neuronal and glial tropism when administered systemically, but transduction is primarily neuronal in neonatal animals and primarily glial in adults [41–43]. Penetration of the intact blood–brain barrier and transduction of brain tissue is limited with other serotypes, including AAVs 1, 2, 5, 6, and 8 [44–47]. See also Chapters 16 and 17 for discussion of systemic AAV administration.
Tropism also differs among AAV serotypes following ocular administration via subretinal injection, which is generally an efficient method for outer retina transduction. AAVs 1, 2, 4, 5, 7, 8, and 9 transduce cells of the retinal pigmented epithelium (RPE) [48–52]. AAVs 1, 2, and 5 transduce these RPE cells with similar efficiency when directly compared [48]. In addition, AAVs 1, 2, 5, 7, 8, and 9 transduce photoreceptors (PRs), while AAV4 does not [48–53]. AAV8 also possesses tropism for ganglion cells and cells of the inner nuclear layer [50, 54], and demonstrates greater tropism for PRs than AAV2 [54]. In nonhuman primate subretinal injections, AAV8 efficiently targets rod PRs [54], while AAV9 is superior for cone PRs and retains tropism for rod PRs [53]. AAV5 has also been shown to target both rod and cone PRs, but has not been compared directly in this setting against other serotypes [55].
Surprisingly, nervous cell tropism can also vary among vector preparations of the same serotype, even when injected under identical conditions. For example, CsCl-purified AAV8 exhibited strong astroglial tropism following intraparenchymal brain injection, while iodixanol-purified AAV8, injected under identical conditions, transduced only neurons [23]. Variability among vector preps or among injection conditions may therefore explain the differences in tropism that are frequently observed among experiments.
As a result of this variability, it is not possible to confidently restrict gene expression to neuronal or glial populations based solely on the capsidserotype. Thus, a cell type-specific promoter should be utilized if astrocyte-, oligodendrocyte-, or neuron-specific transduction is desired. However, most serotypes preferentially transduce neurons following intraparenchymal brain injection, and therefore a pancellular promoter can be used to drive neuronal gene expression, so long as the potential transduction of glia is not problematic. On the other hand, if strong glial expression is desired, a cell type-specific promoter should be utilized.
In some cases it may be desirable for AAV to undergo axonal transport, either to increase the spread of gene transfer, or to retrogradely target a specific subpopulation of projection neurons. For example, anterograde transport of AAV9 injected into the ventral tegmental area of the brain can greatly enhance transgene distribution [34], and retrograde transport of AAV1 injected into muscle or sciatic nerve can specifically label discrete pools of motor neurons [56]. Axonal transport is a fundamental property of AAV vectors, and thus any vector that is endocytosed at high levels by projection neurons is likely to transduce distal brain regions [57, 58]. AAV1 appears to be more effective than AAVs 2, 3, 4, 5, or 6 for retrograde transduction of motor neurons following muscle or sciatic nerve injection [56]. Further, AAVs 1 and 5 demonstrate greater retrograde transduction of brainstem than AAVs 2, 8, or 9 following injection of the transected spinal cord [59]. Within the brain, AAV9 is most frequently observed to undergo axonal transport, and is the recommended choice if distal transduction is desired [21, 23, 34, 36, 57, 60]. In addition, AAVs 1, 8, and rh.10 also undergo axonal transport within the brain and can be used to drive distal transduction [21, 28–30, 34, 57, 60].
3 Modification of the Capsid
To further improve the utility of AAV as a gene therapy vector, research has concentrated on altering capsid properties such as tropism, targeting specificity, and antigenicity. This can be achieved by (1) chemical modification of the capsid; (2) assembling mosaic capsids consisting of subunits from two or more different serotypes; (3) peptide insertion; (4) capsid shuffling; or (5) rational design.
Chemical modifications to improve the tropic properties of adenoviral and lentiviral vectors have resulted in moderate success (reviewed in refs. [61, 62]). Similar strategies have been applied to AAV, although to a lesser extent. Bispecific antibodies capable of binding both AAV2 and αIIbβ3 integrin can increase the transduction of αIIbβ3 integrin-expressing cells by 70-fold [63]. In another study, linkage of biotin-coated AAV to an EGF- streptavidin fusion protein increased the transduction efficiency of EGFR-expressing SKOV3.ip1 cells more than 100-fold [64]. However, despite these promising results, enhancement of AAV tropism has not been achieved in vivo. Chemical capsid modifications can also be used to mask receptor-binding domains on the AAV capsid, de-targeting the virus from its native receptors, allowing infection through alternate receptors, and shielding the capsid from neutralizing antibodies. Indeed, moderate success has been achieved by coating capsids with poly(ethylene) glycol [65], poly-[N-(2-hydroxypropyl) methacrylamide] [66], and α-dicarbonyl compounds [67]. However, chemically modified capsids have yet to be widely used in vivo, and their utility in the CNS remains limited.
AAV capsids are assembled as icosahedral particles from 60 subunits of the VP1, VP2, and VP3 structural proteins. Hybrid capsids are designed to harness the structural similarity among AAVs, combining beneficial properties from two or more different serotypes by co-expressing their capsid proteins during vector production [68]. Hybrid capsids of AAV1 and AAV2 (AAV1/2) can mediate stronger transgene expression in lung and muscle in vivo than either of the parental serotypes alone [69]. Further, AAV1/2 appears to combine the tropism of AAV2 for TH-positive dopamine neurons with the ability of AAV1 to diffuse more widely through brain tissue, mediating strong transduction of dopamine neurons in the substantia nigra, and has been used to model Parkinson’s disease in the rat [70]. Hybrid capsids can also be used to transfer binding affinities from their parental serotypes, such as HSPG binding (AAV2 or AAV3) or mucin binding (AAV4 or AAV5) [71]. However, although the composition of hybrid capsids can be influenced by expressing the parental cap genes at specific ratios, the composition of individual capsids cannot be directly controlled, and undesired capsid arrangements are likely to occur. Furthermore, direct genetic manipulation of the cap gene provides a more precise method by which the properties of different serotypes can be combined, and thus hybrid AAV capsids are rarely utilized.
The earliest successful alterations of AAV tropism by capsid engineering relied on insertion of short peptides into the AAV capsid [72]. Inserted peptides are displayed on the capsid surface and provide affinity for a receptor specifically expressed by the target cell type. Simultaneous disruption of the native capsid tropism can increase the likelihood of specific interaction with the novel target receptor. However, early attempts to provide AAV5 with the ability to bind HSPG indicated that conferring efficient receptor binding does not necessarily confer efficient transduction of the target tissue. Mutant AAV5 virions, despite being able to bind HSPG as efficiently as AAV2, lost their native infectivity and thus did not demonstrate increased tropism for HSPG-expressing cells [73]. In addition to specific insertions of known receptor binding peptides, insertion screens of random peptide libraries have also been utilized. In both cases, it must be ensured that the insertion does not negatively affect vector production, infectivity, or other properties required for gene transfer. Several regions of the capsid are amenable to insertions, including the N-termini of VP1 and VP2, as well as the various loop regions shared by all VP proteins [72, 74–78]. A peptide inserted into the common C-terminal domain shared by all three VP proteins will be displayed on every capsid subunit (60 copies per viral particle), whereas a VP1 or VP2 insertion will only be present in up to 6 or 12 copies per capsid, respectively. This is an important consideration, as the density of receptor-binding peptides on a capsid can affect its tropism [79]. Although initial experiments focused on the insertion of small peptides (5–15 amino acids), later studies have shown that insertions of full length proteins, such as GFP or mCherry, can also be tolerated without loss of virus function [78, 80]. However, despite these promising in vitro proof-of-principle studies, few AAV mutants with enhanced tropism in vivo have been published. To date, improved targeting of skeletal muscle [81, 82], cardiac muscle [83], vasculature [84–86], lung [87, 88], diseased brain endothelial cells [89], retina [90], ovarian cancer cells [91] and breast cancer cells [88] has been reported. Most of these peptide insertions are targeted between amino acids 587 and 588 of AAV2, the region that mediates HSPG binding [73, 92], in order to disrupt the function of this region and de-target AAV2 from its native tropism. The AAV2-7m8 mutant, which was generated via random insertion of a seven amino acid sequence, can efficiently target most retinal cell types following intravitreal injection [90].
In addition to peptide insertion, the development of capsid shuffling [93] and directed evolution [94], discussed in Chapter 11, has generated many promising novel AAV variants. Briefly, cap genes of different AAV serotypes are nuclease digested, mixed together, and randomly reassembled to produce mutated chimeric genomes, which are subsequently selected for a specific function or tropism via directed evolution screening. Capsids with improved transduction of heart [95], lung [96, 97], Müller glia in the retina [98], CNS [99–101], and neural and pluripotent stem cells [102, 103] have been described, with numerous others yet unpublished. Directed evolution was also used to screen the random insertion of short peptides into AAV2 VP3, resulting in the AAV2-7m8 mutant capsid, which is capable of transducing all retinal layers after intravitreal injection [90]. In addition to AAV2-7m8, ShH10 can specifically transduce Müller cells from the vitreous [98].
An improved understanding of AAV structure and biology has enabled researchers to modify vector function by rationally targeting mutations of amino acids on the viral capsid, rather than selecting clones with the desired property from a library of mutants. Some rationally designed mutants combine the desired functions of different serotypes, while others disrupt the domains responsible for unwanted characteristics. The former group includes AAV2i8, a chimera of AAV2 and AAV8 [104], AAV2.5, a chimera of AAV2 and AAV1 [105], and chimeras of AAV1 and AAV6 differing by single amino acid changes [106]. These studies indicate that changing only a small number of amino acids is sufficient to generate capsids with the characteristics of both parental serotypes. In addition, AAV2i8 and AAV2.5 possess unique antigenic properties [104, 105]. Similarly, AAV6.2, a novel vector with improved transduction in mouse airways, was generated via targeted single amino acid changes to AAV6 [107, 108]. Disruption of native AAV properties by targeted amino acid mutations has primarily focused on masking the capsid from neutralizing antibodies, which can inhibit AAV-mediated gene transfer. Several mutations that alter serum antibody recognition and neutralization of AAV2 while retaining normal vector function have been identified [109, 110]. Rational mutations have also been designed to increase transduction efficiency, reducing the vector dose required for clinically relevant transgene expression and avoiding the immune response associated with large viral loads. It is hypothesized that phosphorylation of the AAV capsid leads to ubiquitination and subsequent proteasome-mediated degradation of the vector particle, reducing transduction efficiency [111]. Indeed, mutating several surface exposed tyrosine and threonine residues significantly increased the transduction efficiency of AAVs 2, 5, and 8 [111, 112]. As hypothesized, proteosomal degradation of these capsid mutants was reduced, resulting in increased viral nuclear transport and transgene expression [111, 113, 114]. In addition, novel transduction patterns are observed when AAV2 tyrosine mutants are applied to the mouse retina, in particular following intravitreal injection, which may eliminate the need for surgically challenging subretinal vector administration [115, 116]. Tyrosine mutations of different serotypes also demonstrate improved transduction of mesenchymal stem cells [113] and the mouse brain [117, 118]. A similar strategy to disrupt AAV2 phosphorylation by targeting serine, threonine, or lysine residues can increase liver transduction in mice [119]. Although tyro-sine mutations improve retinal transduction following intravitreal injection, in the only comparison published to date, AAV2-7m8 demonstrated more efficient transduction from the vitreous [90]. Finally, double tyrosine-mutant AAV9 vectors containing AAV3 ITRs and the neuron-specific synapsin promoter appear to possess stronger neuronal tropism than AAV9 in the murine CNS following systemic delivery, although these mutant vectors were not compared directly against AAV9 or other variants [117].
4 Additional Methods to Refine Gene Targeting
The broad natural tropism of the AAVcapsid can be enhanced or made more specific by harnessing cell type-specific promoters. For example, the 1.3 kb CaMK2a promoter can drive transgene expression in glutamatergic excitatory neurons with high specificity [120]. Further, the 1.8 kb neuron-specific enolase promoter [121], the 470 bp human synapsin-1 promoter [120, 122], the 229 bp MeCP2 promoter [123], and the 2 kb herpes simplex virus 1 latency associated transcript promoter [124] can all drive neuron-specific gene expression. The GFAP promoter can drive astrocyte-specific expression, and the myelin basic protein (MBP) promoter can drive oligodendrocyte-specific expression [26, 125]. However, in order for these promoters to be effective, the AAV capsid must possess tropism for the target cell type. For example, AAV4 carrying a CaMK2a promoter is unlikely to drive strong expression within excitatory neurons, as AAV4 does not transduce this cell type efficiently [22]. Similarly, AAV2 carrying a GFAP promoter was found to drive expression primarily in neurons following intra-parenchymal brain injection, likely due to the limited astroglial tropism of AAV2 [121]. See also Chapter 6 for discussion of cell type-specific promoters.
Woodchuck hepatitis virus posttranscriptional regulatory elements (WPREs) are frequently included in the recombinant AAV genome, and can increase the strength of transgene expression [121, 126]. However, WPREs drive greater expression not only in the target cell type, but also in off-target cells that endocytose a lesser number of AAV particles. This can decrease the specificity of an engineered vector or a cell type-specific promoter. Although this effect has not been thoroughly studied, WPRE elements are not recommended when high specificity for a single cell type is desired. WPREs have also been implicated as a contributing or causal factor in oncogenesis in preclinical studies, likely via an ORF within the WPRE [127]. Modified versions that eliminate this ORF have been developed [128].
The injected dose and volume can also influence AAV tropism. Raising the injected dose increases the number of AAV particles that are endocytosed by all cells local to the injection site, driving stronger gene expression within off-target cells. For example, when 1.2 × 1011 genome copies (GC) of AAV1 carrying a human synapsin 1 promoter were intraparenchymally injected, gene expression was highly specific for inhibitory neurons [120]. However, raising the injected dose to 1.7 × 1012 GC resulted in similar levels of gene expression within excitatory and inhibitory neurons, and further raising the dose to 8.4 × 1012 GC resulted in gene expression primarily within excitatory neurons [120]. This is likely due to increased uptake of AAV1 by excitatory neurons at higher injected doses. Decreasing the injected volume while maintaining the injected dose is likely to have a similar effect, as this will apply AAV more focally, driving stronger gene expression within a smaller number of cells. On the other hand, decreasing the injected dose, or applying AAV more diffusely by increasing the injected volume, is likely to increase specificity by reducing the number of vector particles that are endocytosed per cell. Low doses of AAV are therefore recommended when cell-specific expression is desired. If strong or widespread transduction is required, a dose escalation experiment can be performed to identify the injection parameters that result in the strongest gene expression without loss of specificity. See also Chapter 14 for discussion of intraparenchymal injection. Similar findings were observed in the retina with AAV8, as increased dose shifted tropism from RPE alone to both RPE and PRs [54].
AAV tropism can also be modified via the inclusion of microRNA (miRNA) target sites within the AAV genome [129–131]. By utilizing miRNAs that are expressed only in certain cell types, gene expression can be specifically reduced within these target cells. For example, subretinally injected AAV5 typically transduces both RPE cells and PRs, as described in Subheading 2, item 7. However, binding sites for the RPE-specific miR-204 can block AAV5-mediated gene expression in RPE cells, resulting in PR-specific expression [130]. Further, binding sites for the PR-specific miR-124 can block gene expression in PRs, resulting in RPE-specific expression [130]. Thus, gene expression can also be restricted from specific populations via miRNA binding sites within the recombinant AAV genome.
原文阅读:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4993104/
https://jneurodevdisorders.biomedcentral.com/articles/10.1186/s11689-018-9234-0
https://www.cell.com/molecular-therapy-family/methods/fulltext/S2329-0501(19)30011-7
https://www.alacrita.com/whitepapers/adeno-associated-virus-gene-therapy-landscape
https://www.genemedi.net/i/aav-packaging
https://www.frontiersin.org/articles/10.3389/fnana.2019.00093/full
















 Figure 1. Strategic Use to Gapmer-Based Targeting to Effectively Discern lncRNA Functions at the Level of Local Transcriptional Activity Versus at the RNA Level
Figure 1. Strategic Use to Gapmer-Based Targeting to Effectively Discern lncRNA Functions at the Level of Local Transcriptional Activity Versus at the RNA Level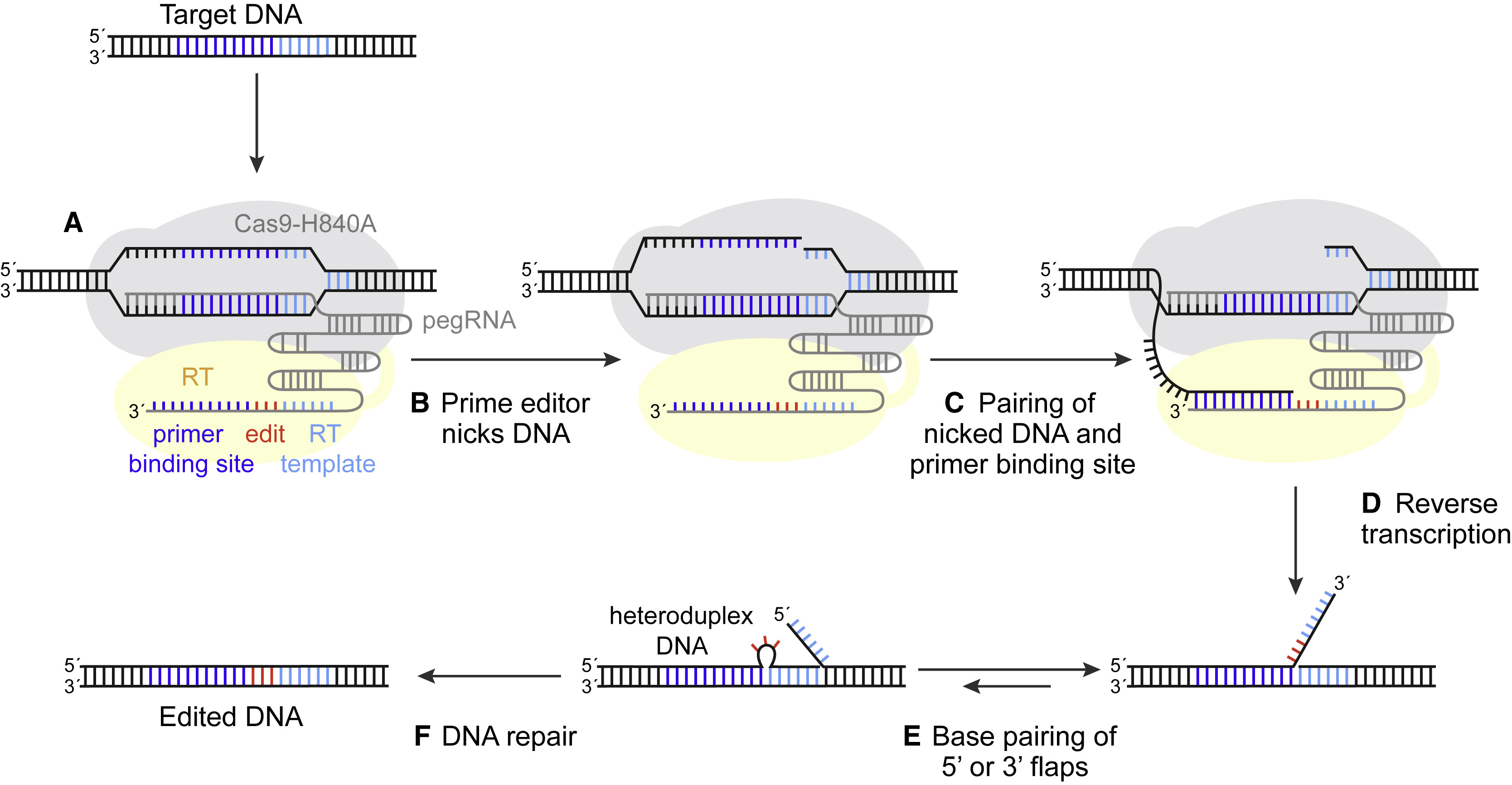 (A) The prime editor (PE) composed of a Cas9-H840A fused to a reverse transcriptase (RT) and pegRNA bind to target DNA.
(A) The prime editor (PE) composed of a Cas9-H840A fused to a reverse transcriptase (RT) and pegRNA bind to target DNA.

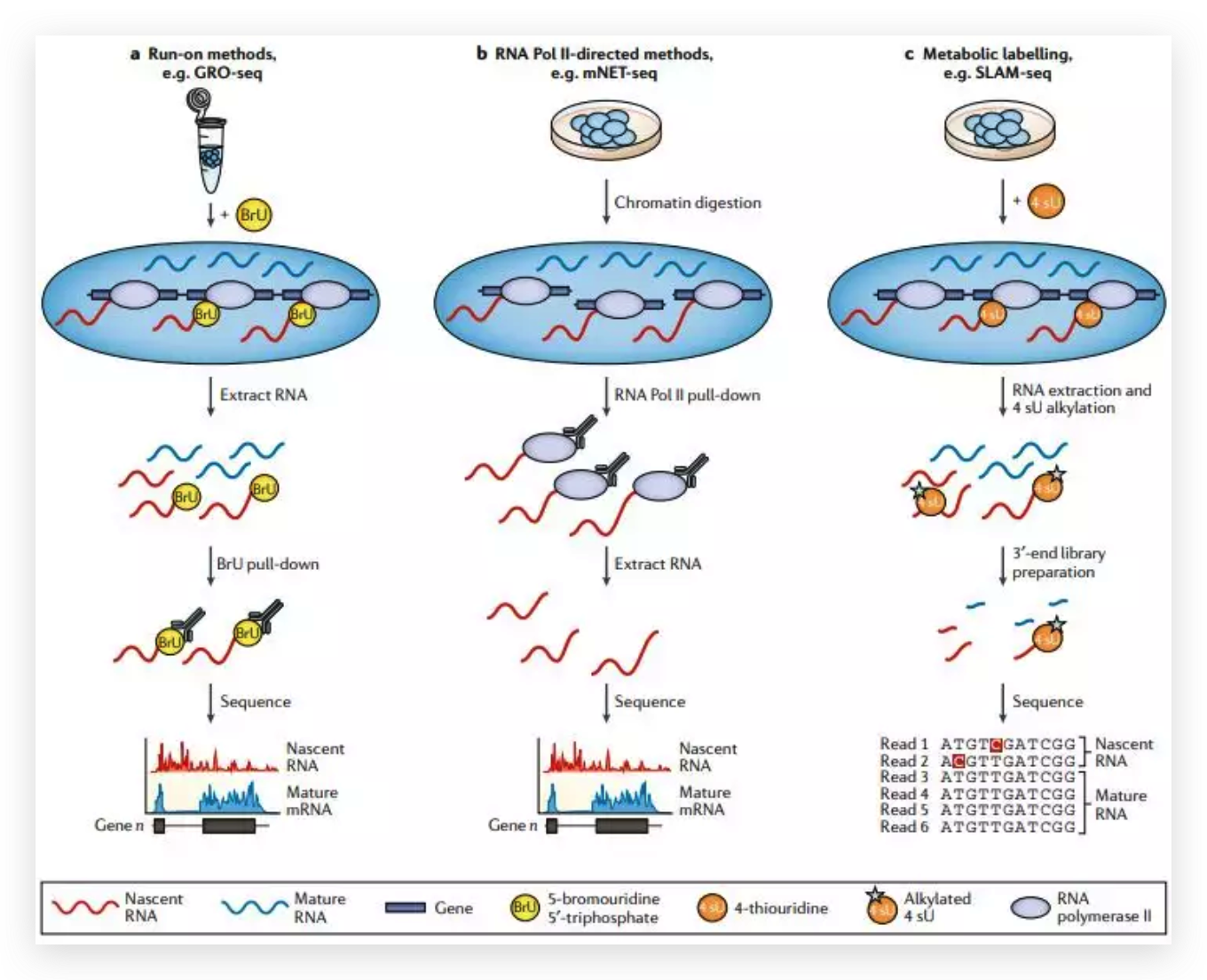
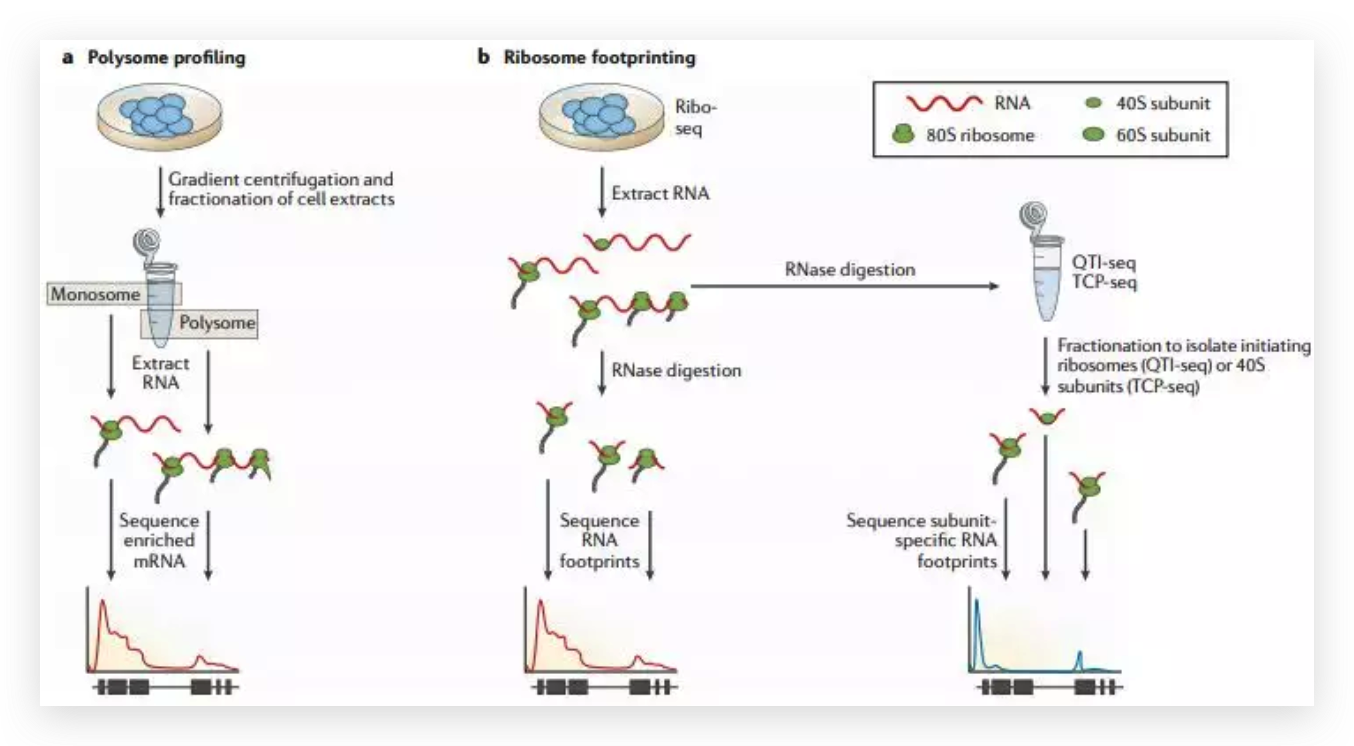
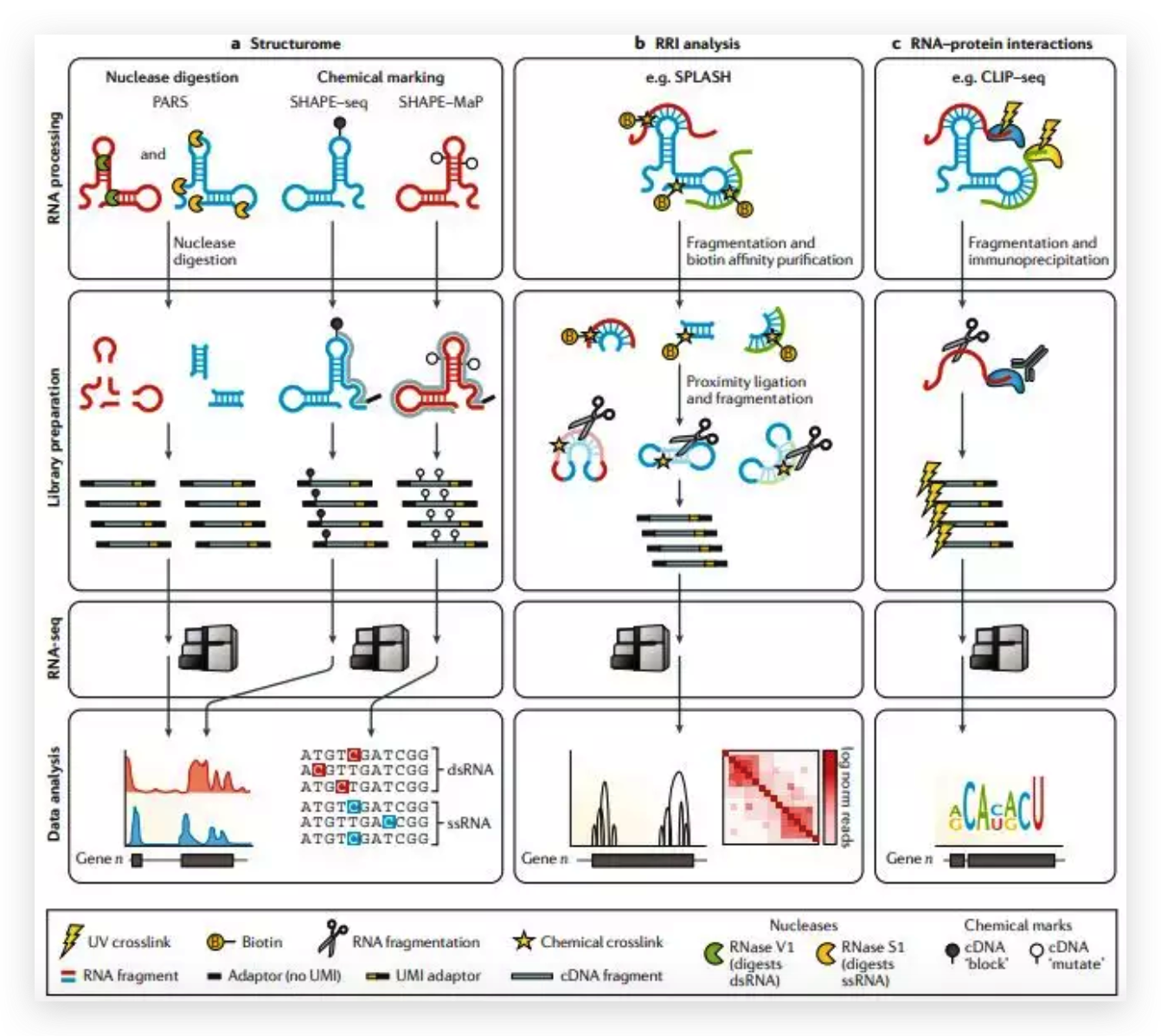
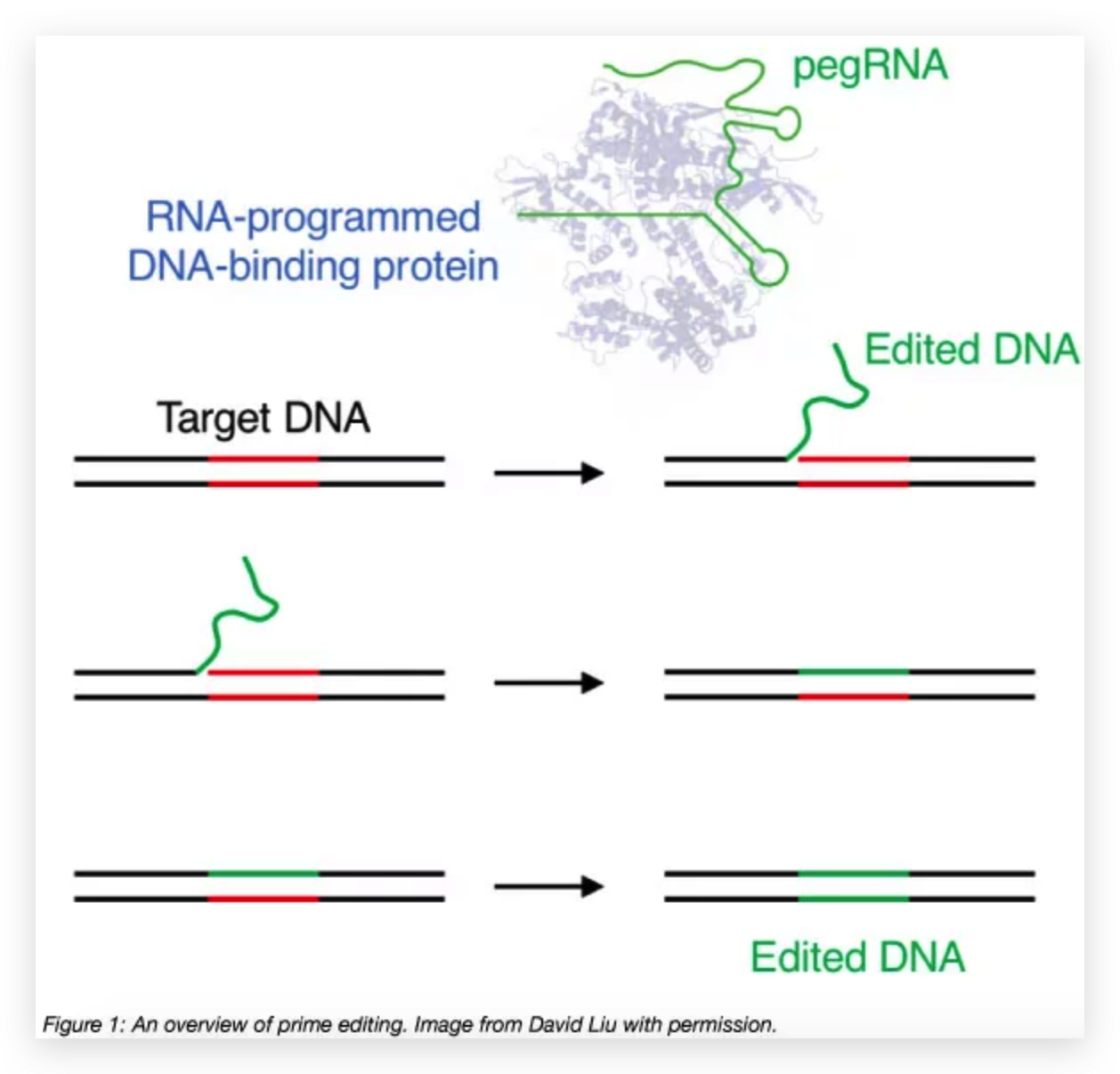
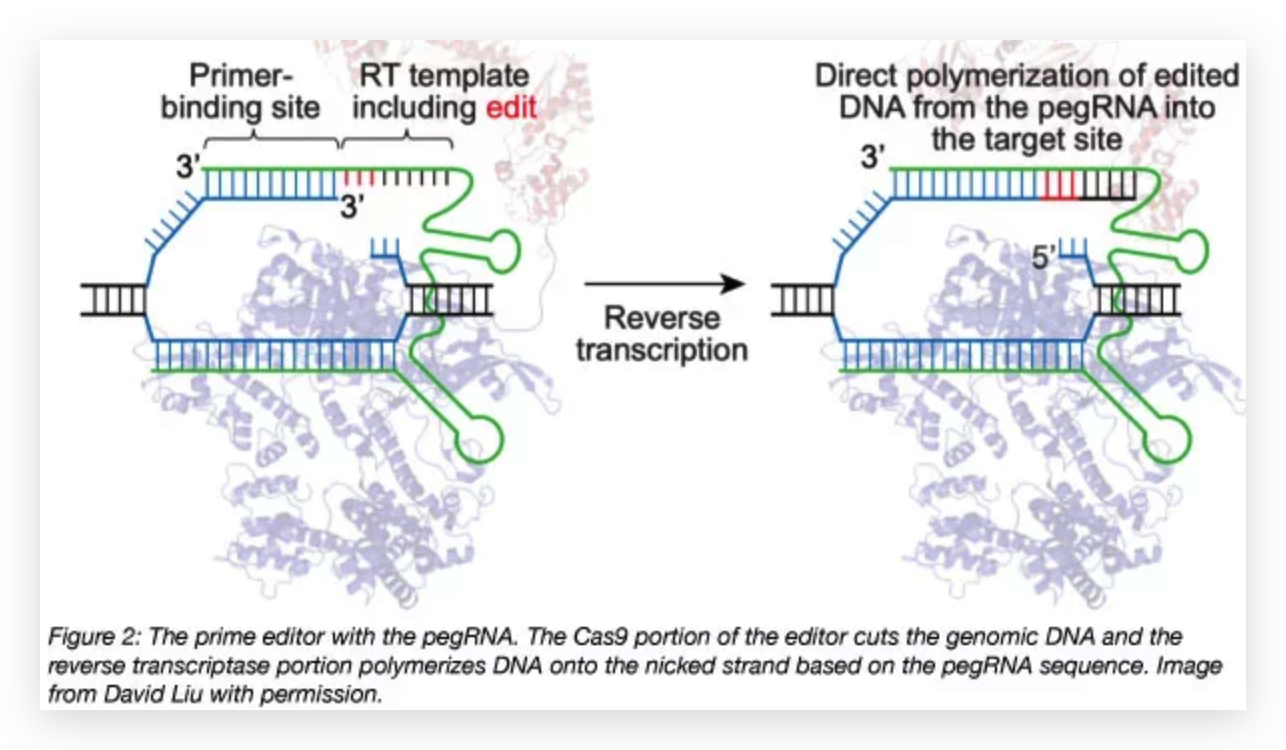
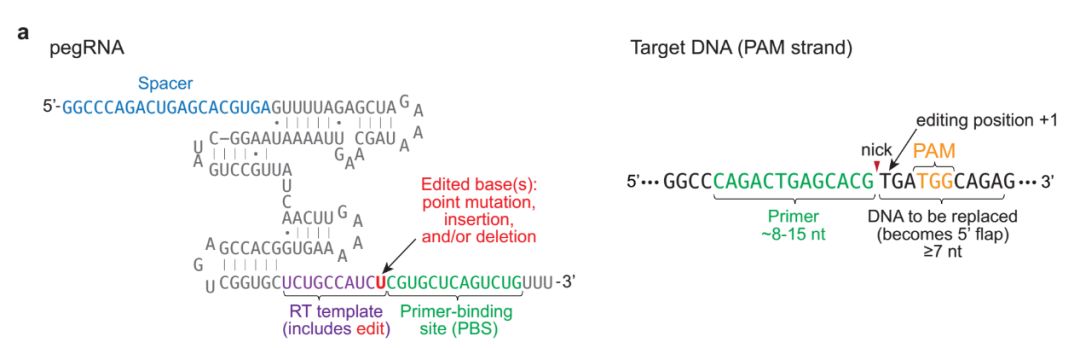
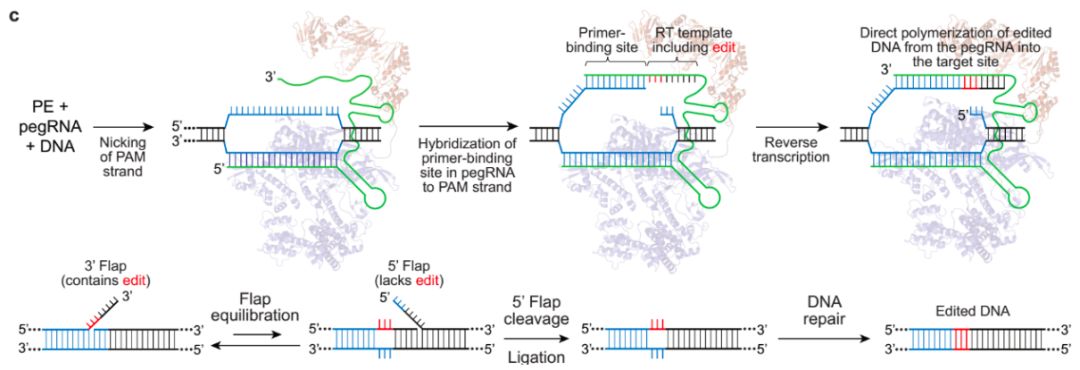
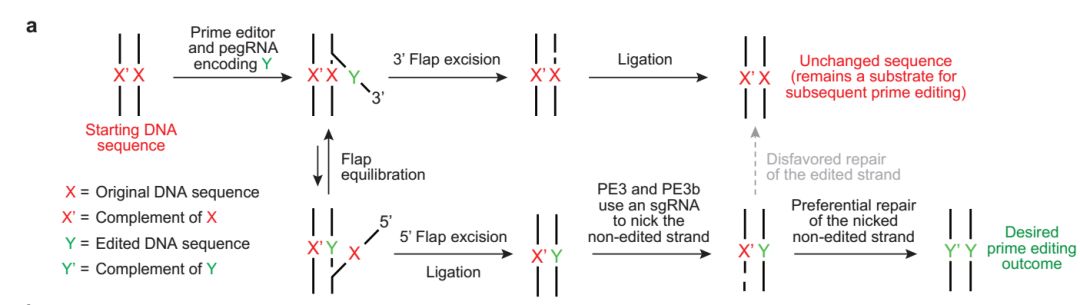
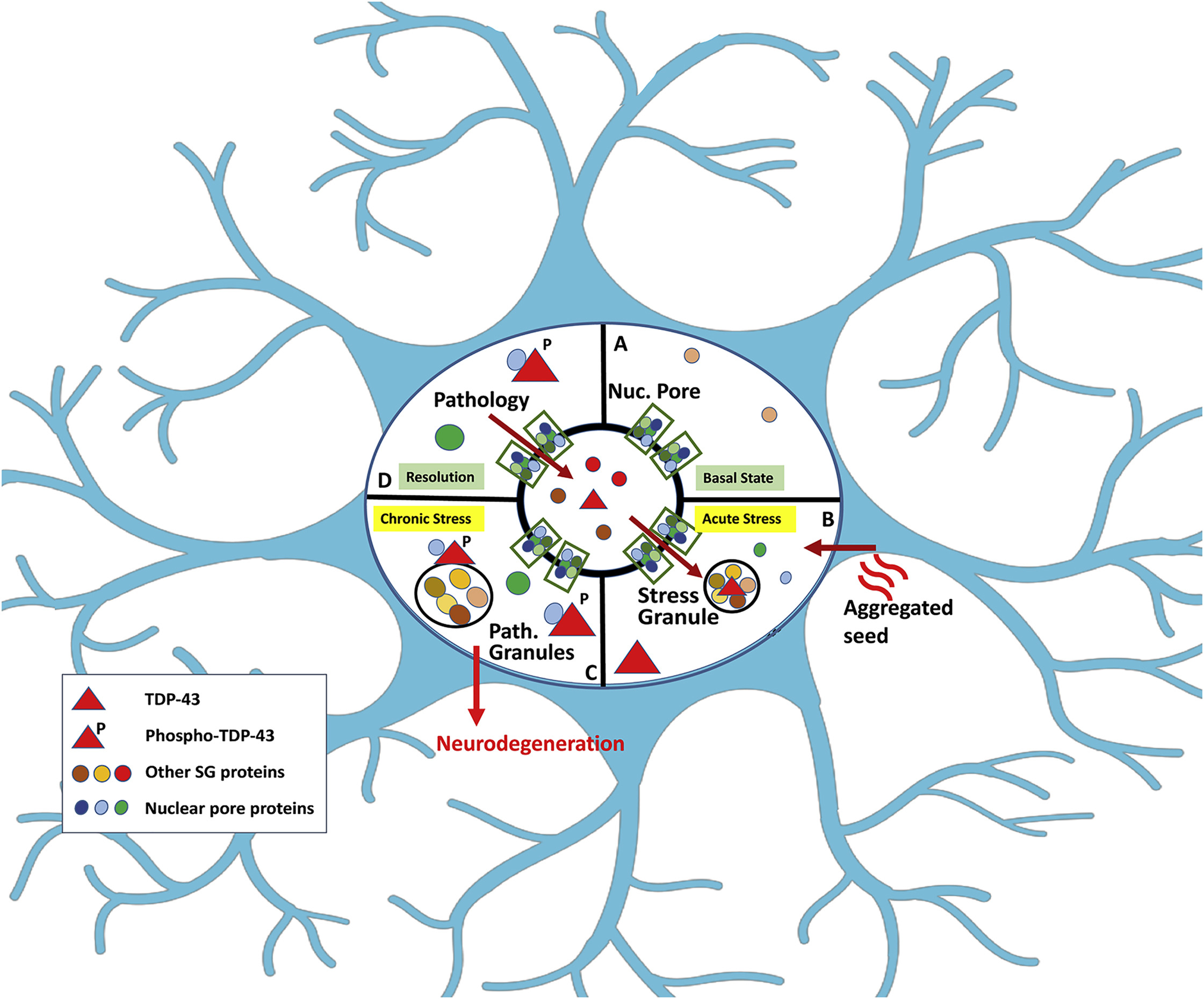
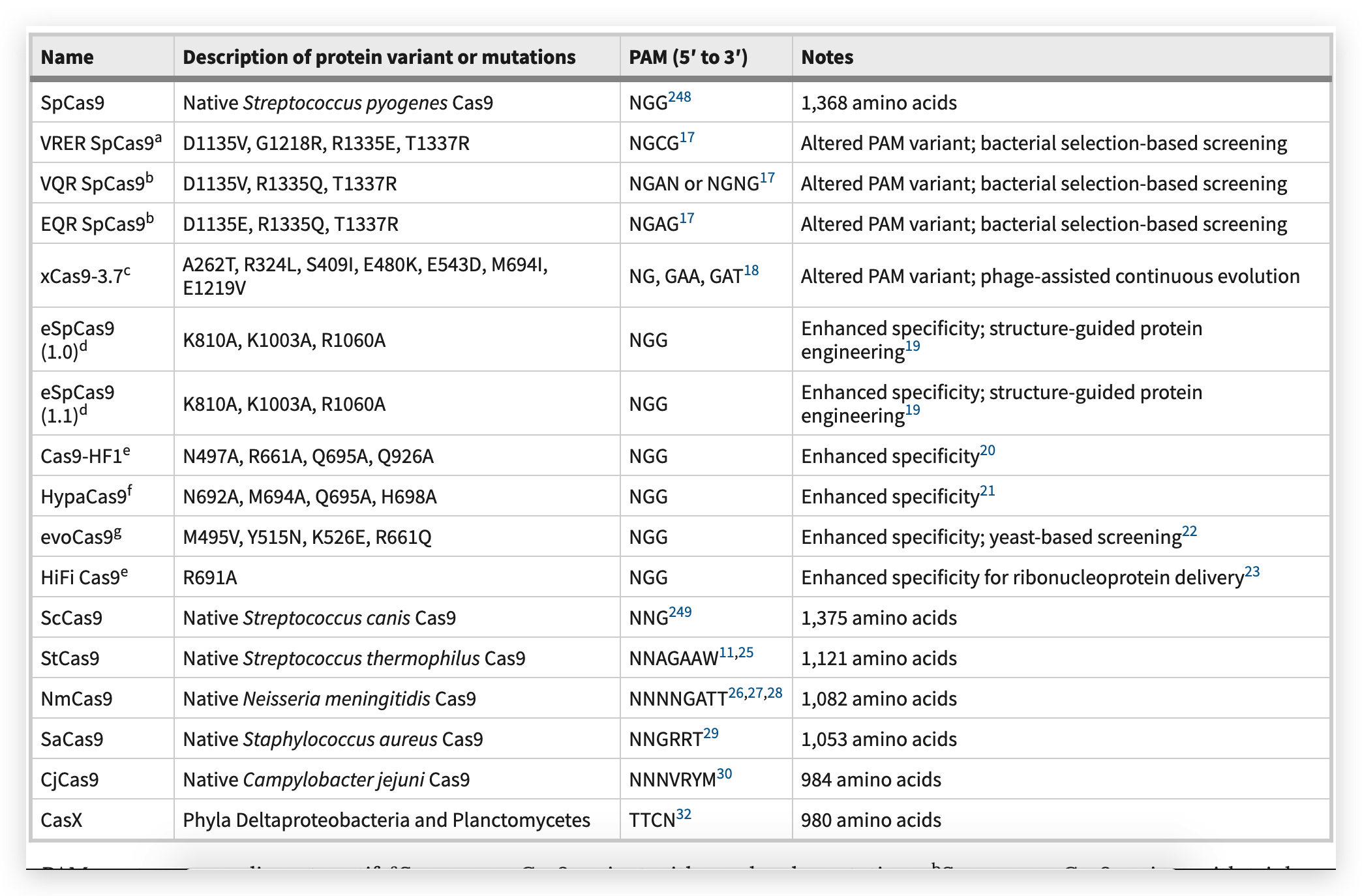
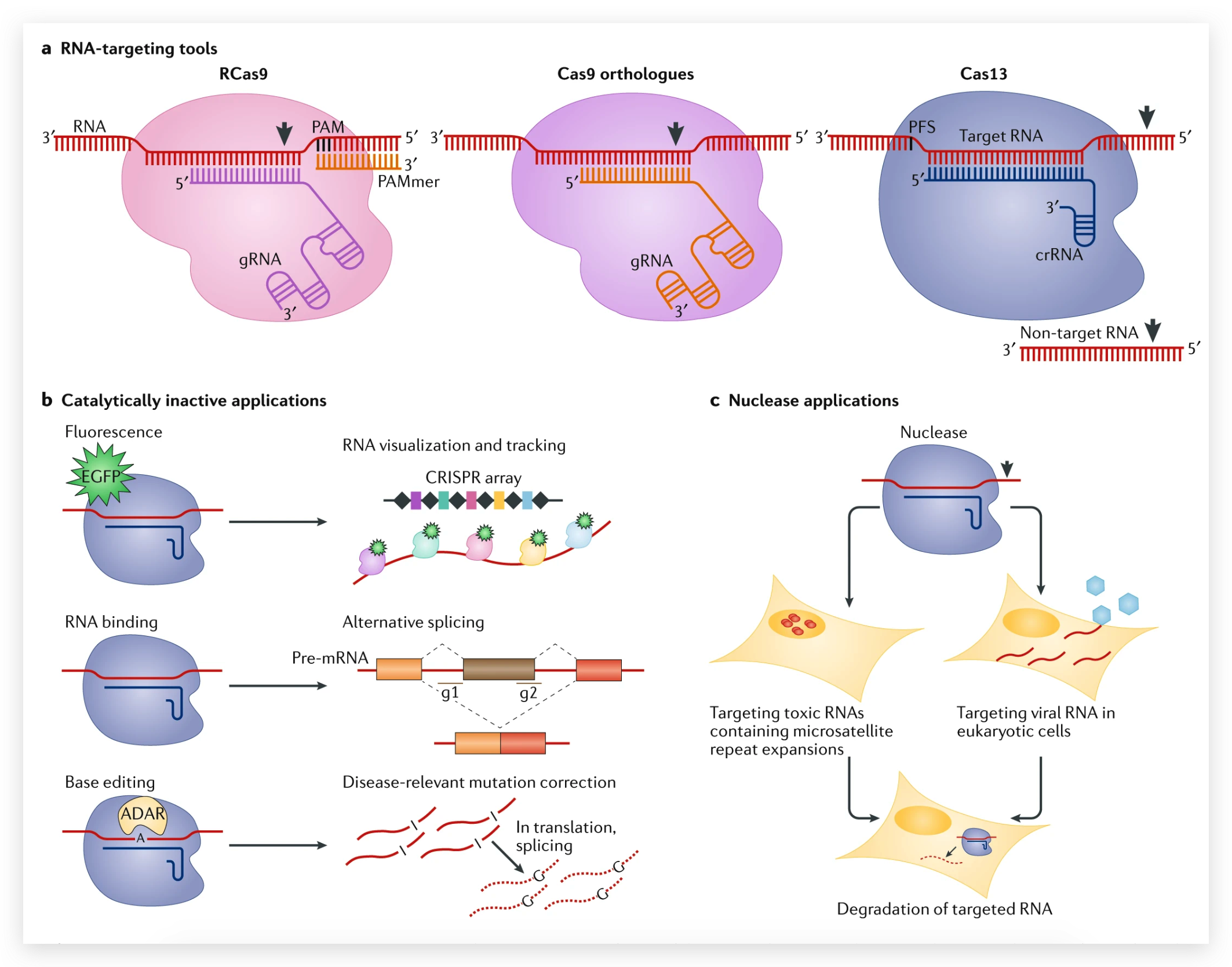
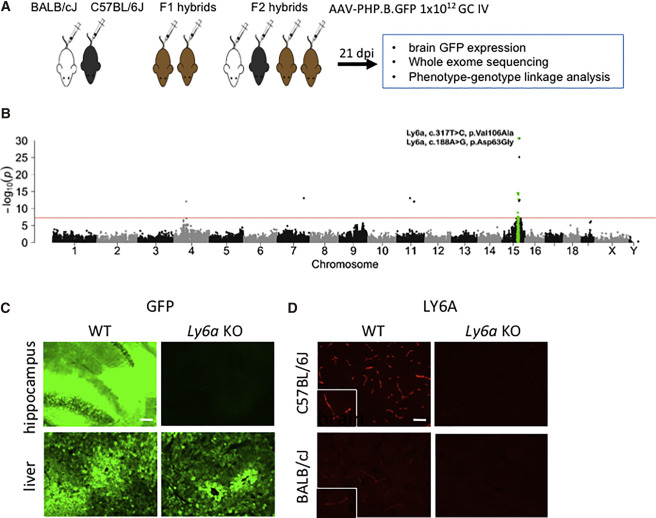


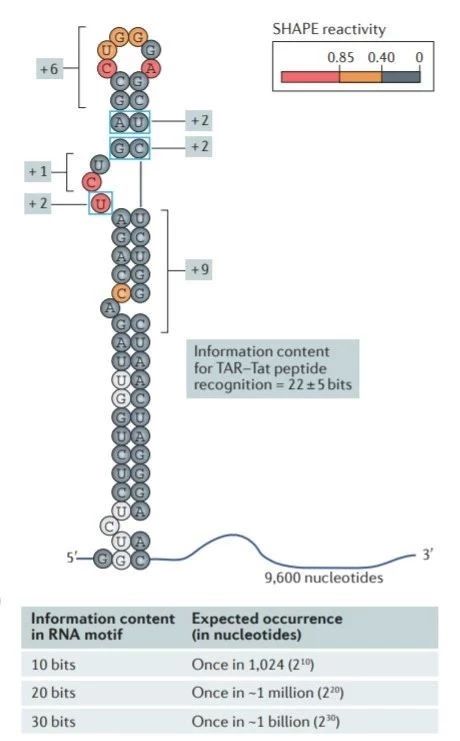
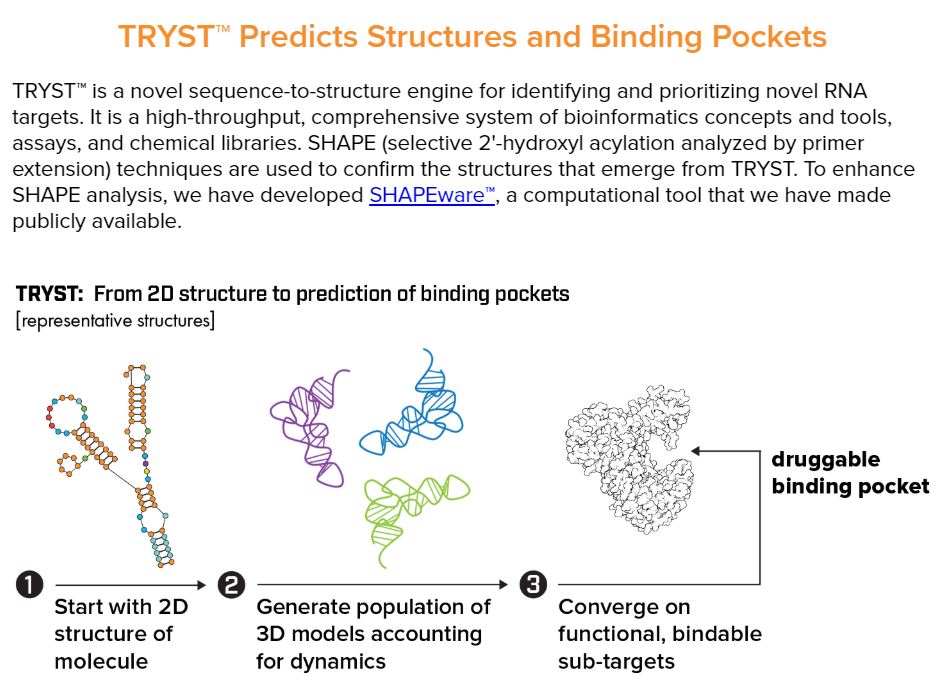

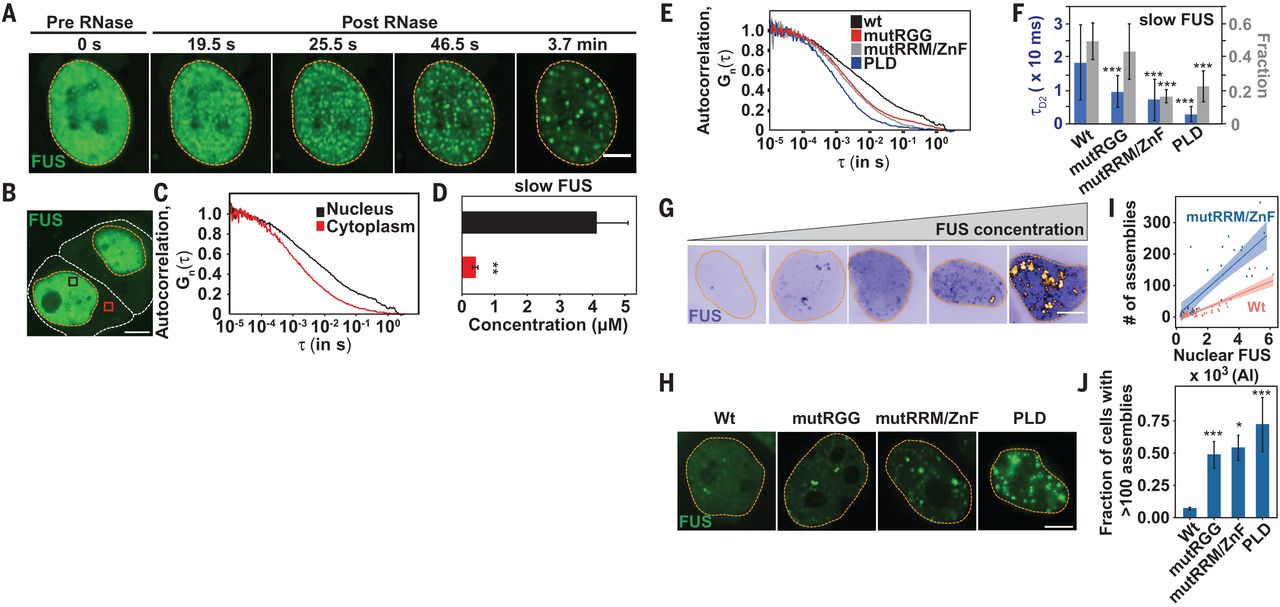

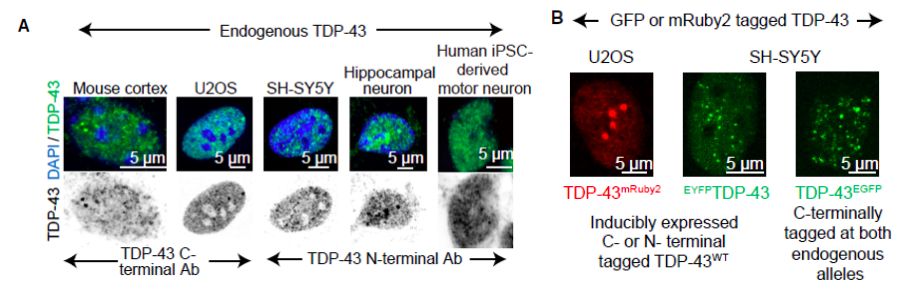
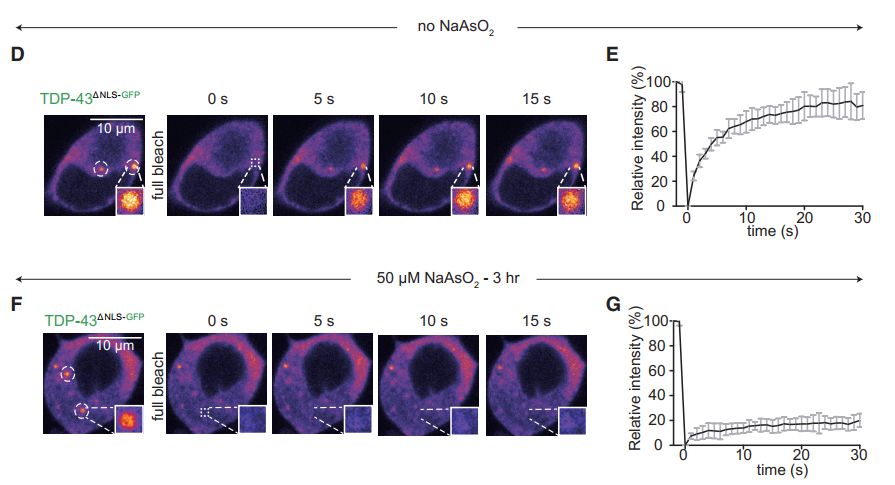
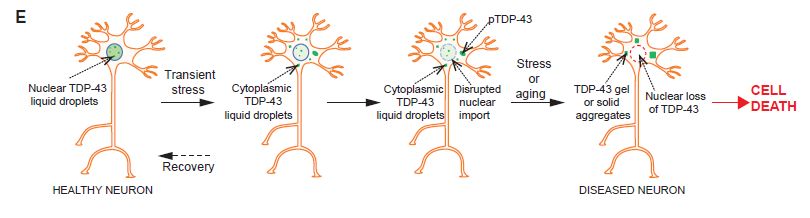
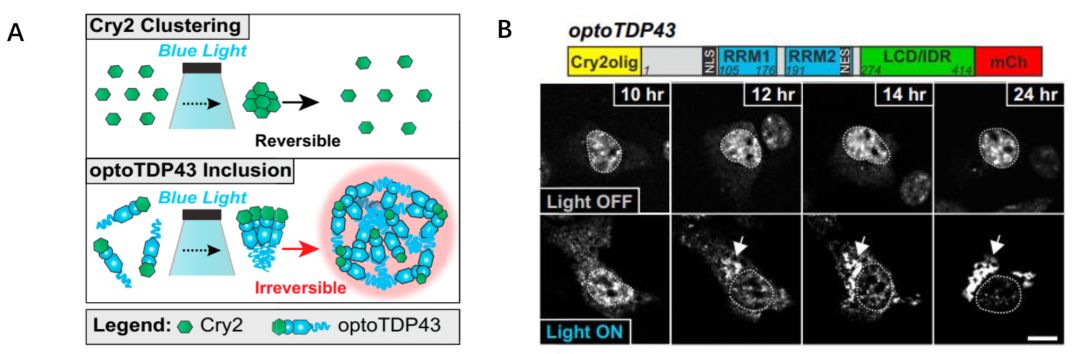
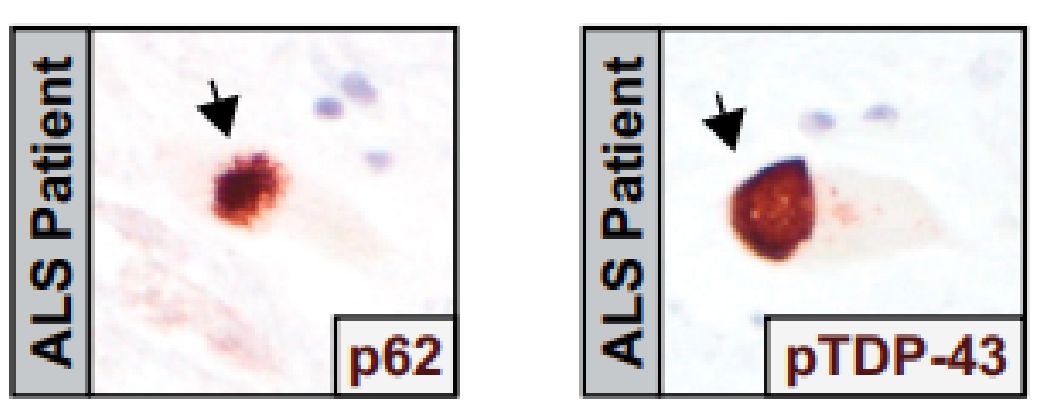
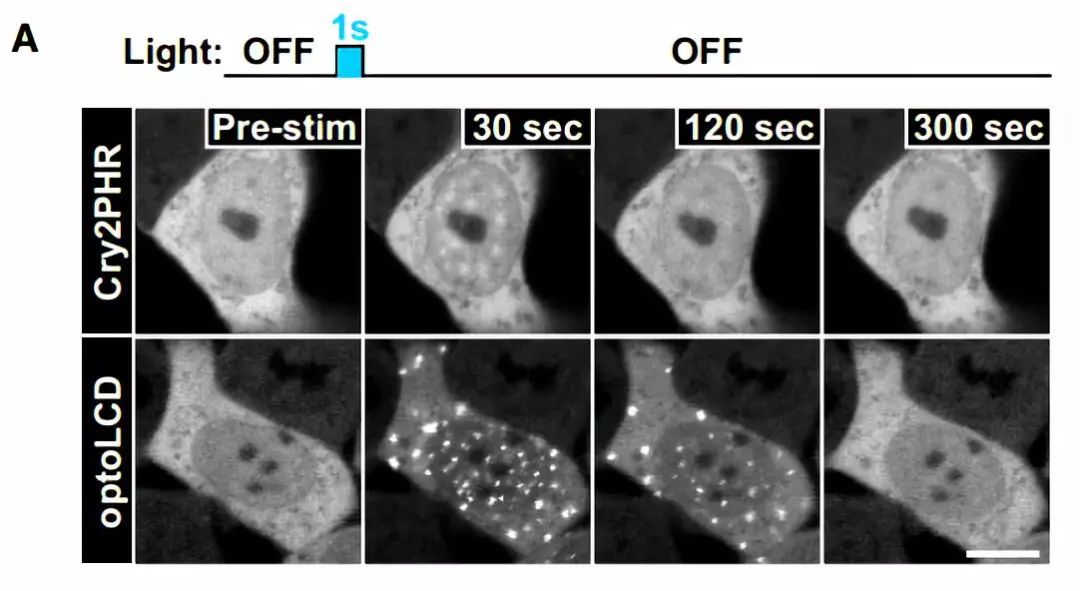 图6 TDP-43的LCD能够在Optodroplet系统中蓝光诱导产生可逆的相分离的现象。
图6 TDP-43的LCD能够在Optodroplet系统中蓝光诱导产生可逆的相分离的现象。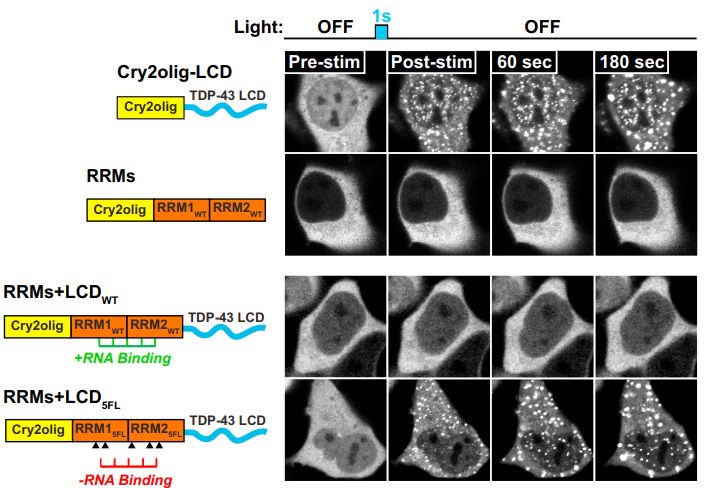

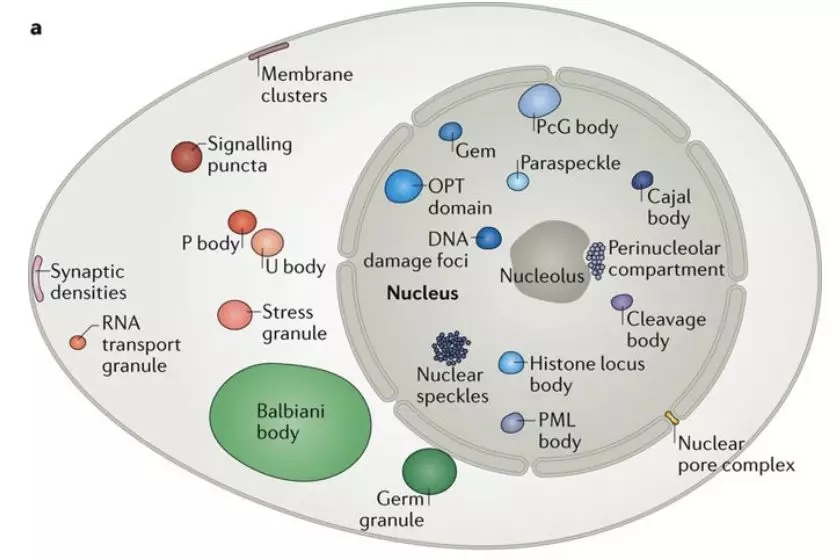

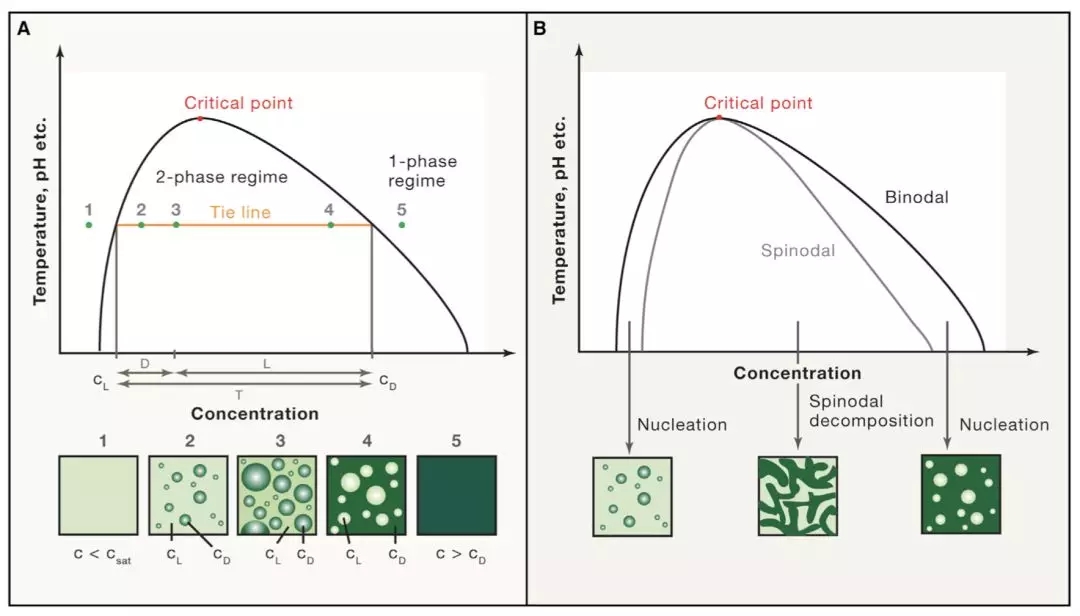

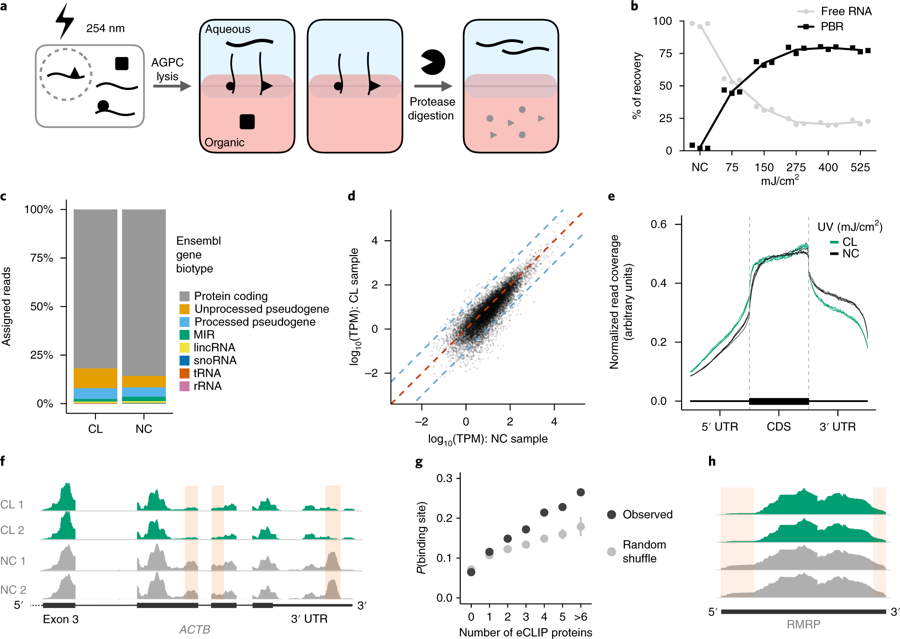 Researchers from the University of Cambridge have developed a new method to capture protein-RNA complexes which is compatible with downstream proteomics or RNA-Seq.
Researchers from the University of Cambridge have developed a new method to capture protein-RNA complexes which is compatible with downstream proteomics or RNA-Seq.