Two articles in Neuron (Volume 102, Issue 2, 17 April 2019) reveal important insights into the potential mechanisms giving rise to the aggregates of TDP-43 that characterize more than 95% of the cases of amyotrophic lateral sclerosis (ALS; Mann et al., 2019, Gasset-Rosa et al., 2019). RNA binding proteins (RBPs) organize their RNA targets by coalescing to create structures called RNA granules. There are many types of RNA granules, but the stress granule (SG) stands out in disease relevance because multiple prior studies suggest that TDP-43 and other disease-linked RBPs co-localize with SGs, which suggests that they might regulate the translational stress response. The linkage of RBPs with RNA granules is conceptually appealing because it places consolidation of RBPs within a physiological pathway that is regulated, providing a model for understanding disease pathogenesis and offering novel targets for therapeutic intervention. However, few groups have observed colocalization of TDP-43 pathology with SG proteins in human brain, which reasonably raises questions about whether TDP-43 pathology truly arises from SG biology.
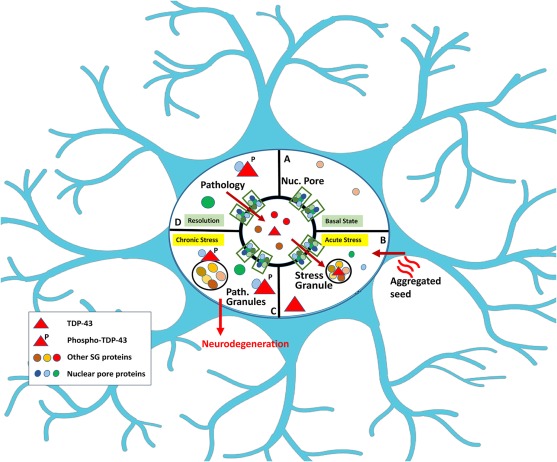
Figure 1. Paths to Formation of Pathological TDP-43 Granules
(A) Basal conditions with TDP-43 in the nucleus.
(B) Stress or pathological seeds induce formation of cytoplasmic TDP-43 granules, as well as some TDP-43 not associated with RNA or stress granules.
(C) The TDP-43 matures into pathological TDP-43 granules.
(D) Neuropathology accrues as unresolvable components accumulate.
The manuscripts by the Donnelly and Cleveland groups provide powerful complement and validation for a model, suggesting that the pathological TDP-43 granules are distinct from SGs, although in some cases appear to transition through an SG intermediate (Mann et al., 2019, Gasset-Rosa et al., 2019). Donnelly’s approach is to investigate TDP-43 aggregation follows the optogenetic approach pioneered by the Brangwynne lab, which used Cry2 to enable optically induced RNA granules composed of FUS and other RBPs (Shin et al., 2017). In the current report, Donnelly’s team added Cry2 to the N terminus of TDP-43 and was able to optically induce TDP-43 oligomers that share similarities with TDP pathology seen in ALS. These granules have properties of gels based on being immobile, unlike classic RNA granules in which the RBPs remain highly mobile as phase separated into liquid droplets; this process is termed liquid-liquid phase separation (LLPS). The optically oligomerized TDP-43 form consolidate that contain phosphorylated TDP-43 and p62, which will be referred to pathological TDP-43 granules. Deletion constructs showed that the low complexity domain (LCD) also possessed the ability to form oligomers; these oligomers are dynamic at first but over 4 h evolve into immobile aggregates that contain TDP-43 phosphorylated at Ser 409/410 (pTDP-43) and p62, which is similar to the neuropathology of ALS.
The twist in the story and physiological insights come from their observations that RNA binding to TDP-43 inhibits formation of the pathological TDP-43 granules. The Donnelly team shows that including RNA recognition motifs (RRMs) prevents the tendency of LCD TDP-43 to aggregate. In addition, while stress induces most TDP-43 to form aggregates associated with SG markers, some stress-induced TDP-43 aggregates contain neither RNA nor SG markers; the heterogeneity of TDP-43 aggregates in stress is something that is commonly observed, but this work provides a useful model for understanding this biology. They emphasize the presence of a SG-independent pathway by showing that optically induced granules of WT TDP-43 also do not co-localize with SGs. This latter observation highlights that TDP-43 can form inclusions through a pathway separate from SGs but also might reflect the ability of the Cry2 system to force the aggregation without the requirement of a physiological pathway to stimulate the process. Finally, they show that a modified oligomer that binds TDP-43 with very high affinity can prevent formation of pathological TDP-43 granules and toxicity, which suggests the presence of crosstalk between the mechanisms producing TDP-43 SGs and pathological TDP-43 granules.
The Cleveland group uses independent approaches yet arrive at similar conclusions. They show that TDP-43 droplets can be induced by sonicated fibrils of aggregates TDP-43 or FUS, but not SOD1. This provides an immediate link to the idea of propagation, which is well accepted from prion diseases, considered quite possible for synucleinopathies and tauopathies but more controversial for TDP-43-opathies (Stewart et al., 2012). A striking aspect of these fibril-induced TDP-43 granules is that they remain as dynamic droplets for up to a month and only form gels with insoluble TDP-43 upon exposure to a stress, such as arsenite. This insoluble TDP-43 contained pTDP-43, suggesting immediate parallels to disease pathology. The pathological transition of TDP-43 observed by both the Cleveland and Donnelly groups is consistent with a recent study from Bonini’s group, who also showed that stress triggered two types of TDP-43 inclusions, one associated with SGs and one that has TDP-43 inclusions containing the pTDP-43 (McGurk et al., 2018). The theme of stress and aggregation also appears in work from Polymenidou’s team, who observed that stress induced a rapid transition of nuclear TDP-43 from soluble oligomers to insoluble, aggregated TDP-43, which they speculated was not functional (Afroz et al., 2017).
These accumulating studies suggest that pathological TDP-43 granules are distinct from SGs yet exhibit crosstalk with SG pathways (Figure 1). For instance, genetic regulators of SGs, such as ataxin-2 and tankyrase 1,2, inhibit the accumulation of TDP-43 pathology in vivo (Becker et al., 2017, McGurk et al., 2018). The SG is a membraneless organelle that forms when RNA translation stalls. SGs are characterized by the presence of mRNA, 40S ribosomal proteins, and particular RNA binding proteins, such as TIA1, G3BP, Caprin, or UBAP2L. Cleveland’s group looks at proteins that co-localize with the TDP-43 granules; importantly, many of their experiments use cell lines in which GFP has been introduced into the endogenous TDP-43 by CRISPR, which avoids overexpression artifacts. Using these lines, they find that almost 100% of the TDP-43 granules co-localized with classic SG markers initially, including RNA, TIA1, G3BP1, or UBAP2L, but after 90 min most of the TDP-43 transitions to granules that no longer co-localized with the SG markers. The observation of SG-independent TDP-43 granules parallels those of the Donnelly and Bonini groups but also highlights that TDP-43 can evolve from a SG into a pathological TDP-43 granule. Indeed, in the Cleveland study, a large fraction of the pathological TDP-43 granules cluster around bona-fide stress granules, which emphasizes their origin and suggests the close relationship with SGs.
This maturation of proteins from SGs, as well as differential effects of varied aggregates, is also observed with tau, where we showed that in hippocampal neuron cultures tau oligomers only associate with SGs transiently during the first hour after exposure to exogenous tau seeds, and then form aggregates not associated with SGs (Jiang et al., 2019). Translating cell culture studies to in vivo results, though, can be tricky. The biological response to tau propagation in vivo appears to be lengthened compared to the response in cultured cells. Propagated oligomeric (but not fibrillar) tau produces SGs whose co-localization with tau remain evident 3 months after injection, which contrasts with the rapid kinetics in cultured cells (Jiang et al., 2019). The similar responses of TDP-43 and tau suggest that the maturation of pathological aggregates might be a generalizable phenomenon, but the relationship between this maturation model and human neuropathology remains a question.
Exogenous fibrils also induce cytoplasmic aggregates composed of multiple nuclear and nuclear pore proteins: is this pathological crossover seeding? Pathological seeding and propagation of disease pathology are thought to contribute significantly to the pathophysiology of prion diseases, tauopathies, and synucleinopathies. The typical case of ALS presents with clinical symptoms in very different regions, but from there, the pathology appears to extend in adjacent areas progressively (Stewart et al., 2012). This pathological duality likely represents what occurs in most neurodegenerative diseases, with the relative proportion of stochastic and propagation events differing among diseases and even among patients.
The study of cellular responses to seeding by exogenous fibrils represents an important aspect of the work by Cleveland’s group. Seeding by sonicated TDP-43 or FUS aggregates, but not SOD1 aggregates, is sufficient to induce TDP-43 pathology and cytoplasmic aggregates of multiple nuclear porins (Nups), as well as significant toxicity. The ability of exogenous FUS aggregates to elicit seeding of cellular TDP-43 aggregates suggests that the occurrence of cross-seeding, which is a phenomenon first noted for some strains of prions in yeast but also seen for α-synuclein and tau. Cross-seeding is further suggested because exogenous TDP-43 or FUS appear able to cross-seed cellular aggregates of nuclear pore proteins, such as Nup62, Nup107, and RanGap1. Interestingly, many of the nuclear pore proteins did not co-localize with TDP-43 aggregates, suggesting that the aggregation occurred through an independent process.
The involvement of nuclear pore proteins is particularly important because their contributions to pathology of multiple type of neurodegenerative disease, including ALS, frontotemporal dementia (FTD), and Alzheimer’s disease (AD), have been increasingly noted since the first observation that C9orf72 repeat expansions disrupt nuclear transport (Zhang et al., 2015). Cleveland’s group found that aggregations of RNA binding proteins, nuclear pore proteins, and possibly other proteins could not be reversed by cycloheximide, which is known to inhibit SG formation. However, it is notable that Lloyd and colleagues found that aggregates of similar proteins that were induced by other stresses could be reversed by other SG inhibitors, such as Isrib (Zhang et al., 2018). This raises the possibility that even the cytoplasmic aggregates of nuclear pore proteins might exhibit crosstalk with the SG pathway.
These studies provide important advances in our knowledge of the mechanisms of formation of pathological aggregates (Figure 1). They highlight a pathological TDP-43 granule that is not a SG but, in some cases, evolves through a SG and in other cases evolves independently of SGs. Future studies will need to elucidate the relative proportion of pathological TDP-43 aggregates that accumulate through each pathway in patients with ALS.
REFERENCES
Afroz, T., Hock, E.M., Ernst, P., Foglieni, C., Jambeau, M., Gilhespy, L.A.B., Laferriere, F., Maniecka, Z., Plu€ckthun, A., Mittl, P., et al. (2017). Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 8, 45.
Becker, L.A., Huang, B., Bieri, G., Ma, R., Knowles, D.A., Jafar-Nejad, P., Messing, J., Kim, H.J., Soriano, A., Auburger, G., et al. (2017). Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544, 367–371.
Gasset-Rosa, F., Lu, S., Yu, H., Chen, C., Melamed, Z., Guo, L., Shorter, J., Da Cruz, S., and Cleveland, D.W. (2019). Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102, this issue, 339–357.
Jiang, L., Ash, P.E.A., Maziuk, B.F., Ballance, H.I., Boudeau, S., Abdullatif, A.A., Orlando, M., Petrucelli, L., Ikezu, T., and Wolozin, B. (2019). TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol. 137, 259–277.
Mann, J.R., Gleixner, A.M., Mauna, J.C., Gomes, E., DeChellis-Marks, M.R., Needham, P.G., Copley, K.E., Hurtle, B., Portz, B., Pyles, N.J., et al. (2019). RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 102, this issue, 321–338.
McGurk, L., Gomes, E., Guo, L., Mojsilovic- Petrovic, J., Tran, V., Kalb, R.G., Shorter, J., and Bonini, N.M. (2018). Poly(ADP-ribose) prevents pathological phase separation of TDP-43 by pro- moting liquid demixing and stress granule localiza- tion. Mol. Cell 71, 703–717.e9.
Shin, Y., Berry, J., Pannucci, N., Haataja, M.P., Toettcher, J.E., and Brangwynne, C.P. (2017). Spatiotemporal control of intracellular phase tran- sitions using light-activated optoDroplets. Cell 168, 159–171.e14.
Stewart, H., Rutherford, N.J., Briemberg, H., Krieger, C., Cashman, N., Fabros, M., Baker, M., Fok, A., DeJesus-Hernandez, M., Eisen, A., et al. (2012). Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol. 123, 409–417.
Zhang, K., Donnelly, C.J., Haeusler, A.R., Grima, J.C., Machamer, J.B., Steinwald, P., Daley, E.L., Miller, S.J., Cunningham, K.M., Vidensky, S., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61.
Zhang, K., Daigle, J.G., Cunningham, K.M., Coyne, A.N., Ruan, K., Grima, J.C., Bowen, K.E., Wadhwa, H., Yang, P., Rigo, F., et al. (2018). Stress granule assembly disrupts nucleocytoplasmic transport. Cell 173, 958–971.e17.
