Novel targeted degrader concepts are appearing almost daily. In September, Genentech researchers unveiled a cancer-targeting strategy that uses bispecific antibodies to induce degradation of cell membrane proteins — the latest addition to the fast-growing family of targeted degraders. The very next day, University of California, San Francisco biochemist James Wells published on bispecific antibodies that link cytokine receptors to extracellular and membrane protein targets. (EpiBiologics has licensed the technology.) These and other degraders take different roads, but the destination is the same: the lysosome.
The prototype degrader is the PROTAC, or proteolysis-targeting chimera, which eliminates target proteins by linking them to an E3 ubiquitin ligase for destruction in the proteasome. But PROTACs can’t degrade many categories of disease target, so academic and industry scientists are extending the degrader concept to a diverse set of new targets by exploiting the cell’s other main recycling center, the lysosome. These bifunctional small molecules have the potential to vastly enlarge the degrader space, but as they are so new, the field is still figuring out how best to select, optimize and deploy them, and how to minimize their risks.
Targeted degraders in general can eliminate targets that are otherwise undruggable, because they need only bind to work, not inhibit, and, unlike small-molecule inhibitors, they remove the target in its entirety. First described in 2001, PROTACs emerged as a viable drug modality in 2015, when Craig Crews’ group at Yale described fully small-molecule catalytic PROTACs with efficacy at nanomolar concentrations. Developing PROTACS now commands the efforts of about nine biotechs and numerous pharmas, with at least a dozen clinical trials in progress. But PROTACs can degrade only intracellular proteins and a subset of membrane proteins with suitable intracellular domains. Many other potential drug targets remain inaccessible, including secreted proteins and many integral membrane and membrane-associated proteins, as well as protein aggregates, lipids and whole organelles.
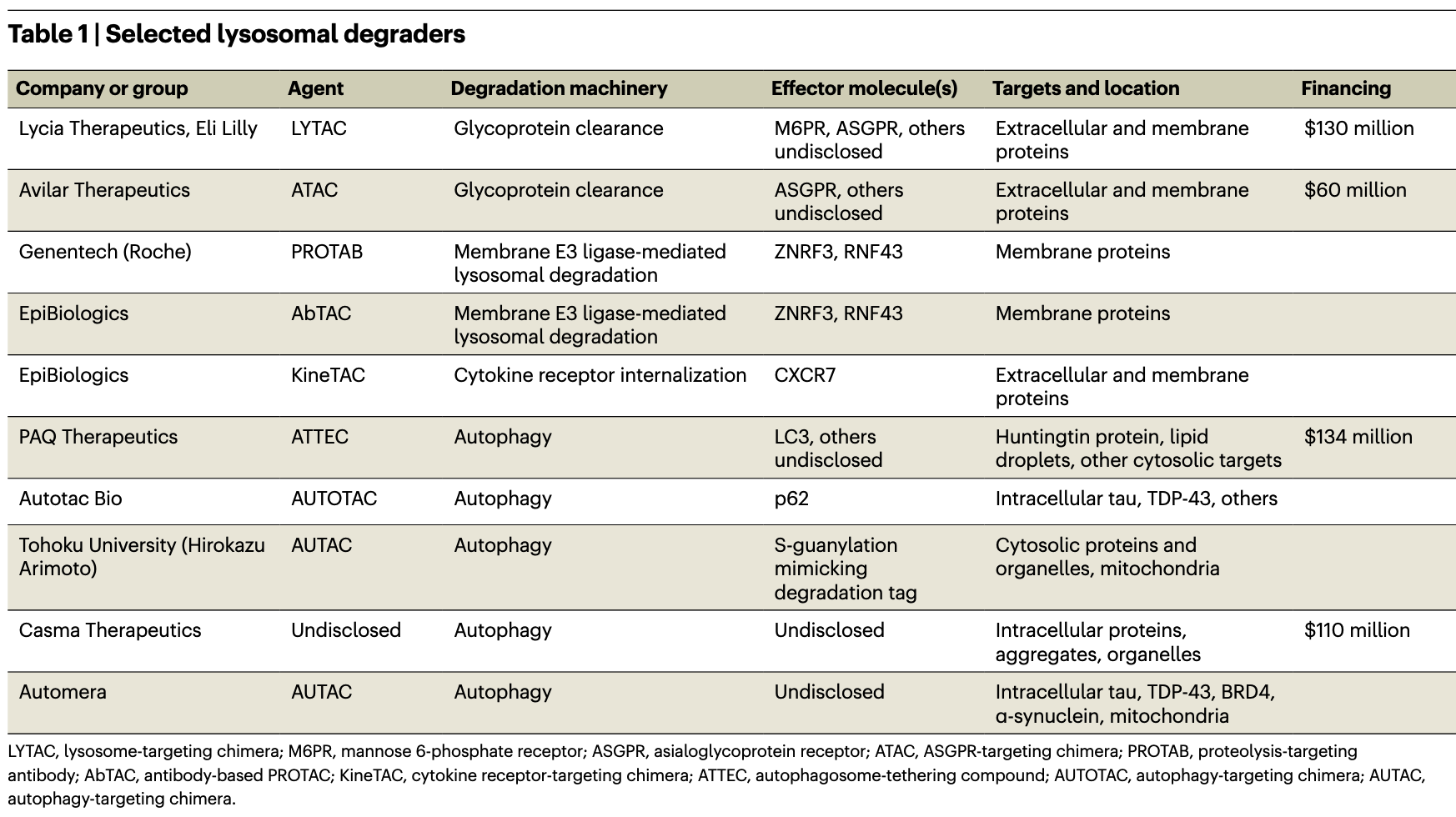
The new generation of degraders avoids the ubiquitin–proteasome system entirely and instead harnesses cellular pathways for delivering cargo to the lysosome for degradation. LYTACs, or lysosome-targeting chimeras, and ATACs, or ASGPR (asialoglycoprotein receptor)-targeting chimeras, link an extracellular domain of a target molecule to a cell-surface receptor that shuttles it, by endocytosis, to lysosomes. Genentech’s PROTABs, or proteolysis-targeting antibodies, and EpiBiologics’ AbTACs, or antibody-based PROTACs, hijack cell membrane ubiquitin ligases that also direct targets to lysosomes, and EpiBiologics’ KineTACs degrade extracellular and membrane proteins by tethering them to cytokine receptors for cell internalization. Several other degraders exploit natural autophagy pathways, in which specialized vacuoles engulf protein aggregates and complexes, or damaged organelles, for lysosomal delivery.
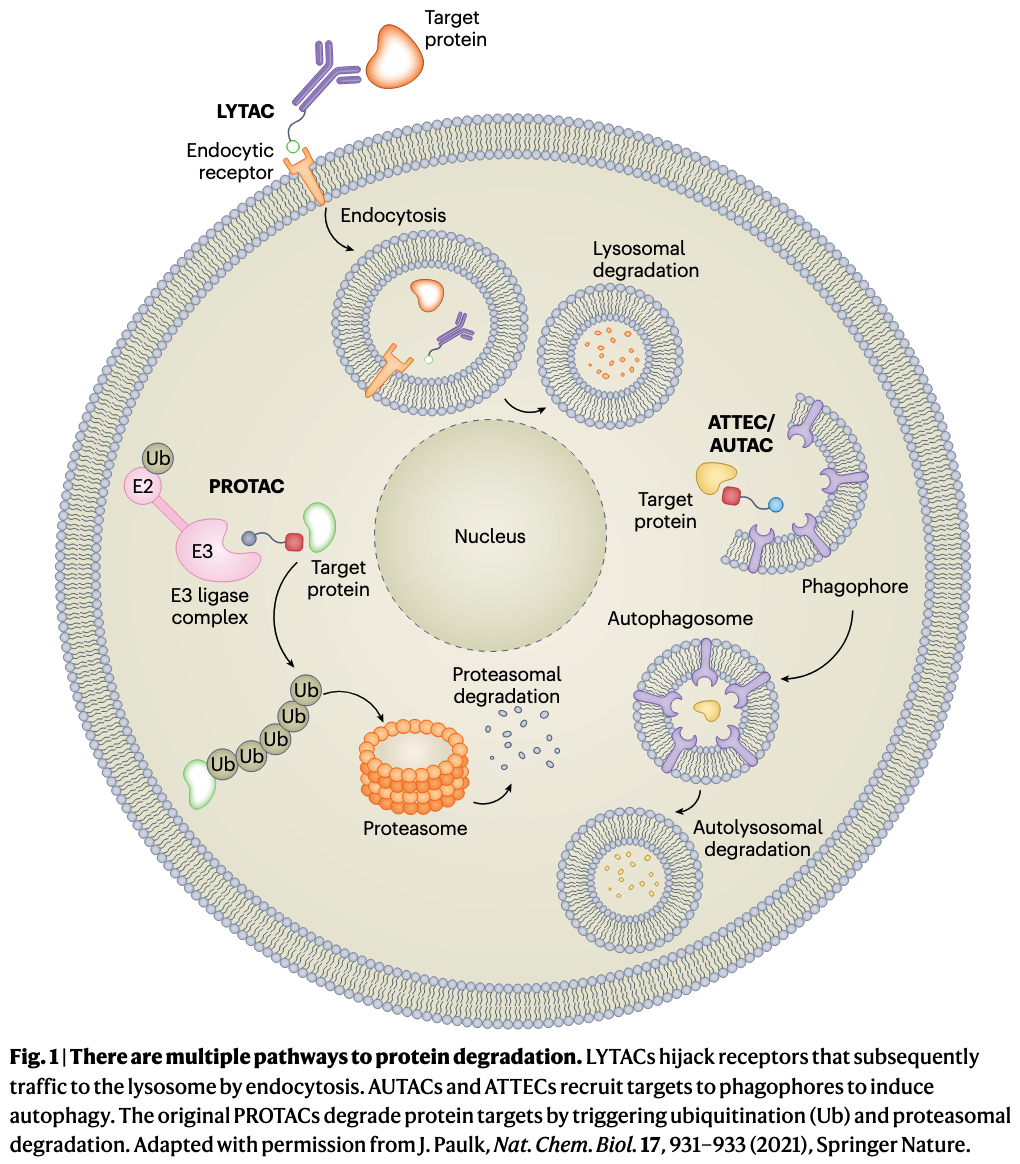
The field of lysosomal degraders is very young but is quickly growing. Besides LYTACs and the other extracellular protein degraders, at least five different autophagy-based degraders are in development (Table 1). At last count, five start-up companies have emerged from stealth mode, with over $434 million dollars invested. But while PROTACs have validated the degrader concept, lysosomal degraders are still untested in patients, and they face challenges and uncertainties as they advance to the clinic. These include oral delivery of bulky degrader molecules, the risk of off-target degradation, the potential to overload lysosomes, and the possibility that hijacking these pathways will disrupt cellular proteostasis.
Garbage in
LYTACs are the most visible new degrader modality. They can theoretically degrade most extracellular and membrane proteins, which together make up about 40% of the proteome. The first LYTACs originated in the Stanford lab of glycobiologist Carolyn Bertozzi, where Stanford University chemical biologist Steven Banik, then a postdoc, was looking for ways to eliminate proteins that crosslink the glycocalyx (the dense sugar-rich layer surrounding cells), which helps drive cancers. The Bertozzi lab had developed artificial glycoprotein polymers, and Banik planned to attach them to glycan-binding disease-related proteins and remove them, by shedding, from the cell surface. But eventually the duo hit on the idea of harnessing the cell’s natural machinery with PROTAC-like bifunctional molecules that operate in the extracellular space and, in combination with membrane-bound transporter molecules, grab extracellular proteins and shuttle them to the lysosome, where they are enzymatically digested (Fig. 1).
The initial LYTAC paper from the Bertozzi lab reported degradation of several therapeutically important proteins — the extracellular protein ApoE4 and membrane proteins EGFR, CD71 and PD-L1 — by linking them to the mannose 6-phosphate receptor, which internalizes and carries glycoproteins to the lysosome. Degrading membrane proteins was a feat because LYTACs must compete with these proteins’ normal internalization machinery. Banik, Bertozzi and colleagues later showed that degraders that used a second internalizing receptor, ASGPR, also worked, in this case in a tissue-specific manner. (Avilar Therapeutics’ ATACs are similar.)
In 2019 Bertozzi founded Lycia Therapeutics to develop and commercialize LYTACs. A $50 million A round followed, and in August of 2021 Lycia signed a research collaboration and licensing agreement with Eli Lilly. The companies have yet to disclose any targets or disease indications, except for Lilly’s focus on immunology and pain. “We’re looking for areas of biology and targets where antagonism has failed,” says Lycia CSO Steve Staben: “things like difficult-to-drug protein aggregates, immune complexes, some of these recalcitrant and ligand-independent membrane targets.” Autoantibodies are also attractive. Other possible targets include membrane proteins with multiple functions and interacting partners, making it necessary to completely remove the protein. Many receptor tyrosine kinases fall into this category, including cancer drivers EGFR, HER2 and FGFR. “A single degrader approach for one of those may be more efficacious than simple kinase domain inhibition or blocking natural ligand binding,” says Staben.
LYTACs enjoy more design flexibility than PROTACs because they don’t have to be cell permeant. Small molecules, peptides, monovalent antibodies, bispecific antibodies — almost anything works if it can bring the target to the receptor. “It’s important for us to have multiple modalities to target these extracellular proteins,” says Lycia president and CEO Aetna Wun Trombley. “You can imagine a fully small-molecule LYTAC that has quite a different pharmacological profile than a fully biologic LYTAC. And so, depending on the situation, it may be more beneficial to apply one of those modalities versus another.”
LYTACs are also relatively straightforward to design. In contrast, PROTACs must bind both targets in a ternary complex that enables the ligase to transfer ubiquitins to the target protein’s lysine residues. The lysines must be oriented properly for ubiquitin binding, and ubiquitination doesn’t always lead to proteasomal degradation. The rules for these interactions remain largely unknown, so drug discovery is empirical and expensive. LYTACs are more forgiving. “You just need to induce proximity transiently to the internalizing receptor,” says Staben. Once achieved, “there’s no need to maintain that ternary complex formation necessarily, there’s no absolute requirement for the suitable presentation of lysines.”
But unlike PROTACs, first-generation LYTACs do not act catalytically, so they may not be able to clear highly abundant proteins. Lycia isn’t ruling out catalysis in the future. “It’s more an engineering feat at this point,” says Banik. “We need to find something that’s going to let go of cargo in the lysosome while it itself can be recycled back to the cell surface.”
LYTAC toxicity, at this early stage, is unknown, but the general approach seems safe because it employs the cell’s normal endocytic machinery. However, the endosomal–lysosomal system is damaged in many diseases and chronic conditions, including neurodegenerative diseases, and the LYTAC cargo burden could make things worse. For example, impaired cargo degradation in lysosomes can lead to lysosomal membrane permeabilization and cathepsin release into the cytosol, inducing either apoptotic or necrotic cell death. “How a cell turns over its proteins is something that I think disease mechanisms tend to alter,” acknowledges Banik. However, “we have at least done initial profiling work to show that LYTACs themselves don’t really cause lysosomal damage in any way.”
Still, Lycia doesn’t want to overload sick lysosomes. “The different disease indications we’re looking at are not known to be associated with dysregulated lysosomal function,” Staben says. And selecting internalizing receptors with specific tissue expression should avoid any problems if issues do arise. “So it may be possible even in some of these patient populations that have lysosomal function changes, that we can avoid hypothetical on-target mechanism-driven toxicity.”
Avilar CEO Daniel Grau says the company hasn’t seen any safety issues in its preclinical studies, including testing in non-human primates. And, like Lycia, Avilar is staying away from diseases with known lysosomal damage or capacity issues. But degraders, by monopolizing the ASGPR (or any internalizing receptor), could theoretically block normal degradation and cause side effects. Grau cites a knockout study showing no reduction in plasma glycoprotein clearance in mice, suggesting that redundant systems are compensating. “There are a lot of backup pathways to degrade proteins,” says Avilar CSO Effie Tozzo. “Also … we’re definitely not saturating the receptor with our ATACs in our pipeline programs.” So degraders shouldn’t affect normal protein clearance. But, until safety is proven in the clinic, such interference remains a concern.
Both Lycia and Avilar are exploring multiple internalizing receptors, but Avilar has staked out a strong chemistry position with ASGPR. “They’ve disclosed quite a bit in terms of optimizing ligands for that receptor,” says Casma Therapeutics CSO Leon Murphy. “It’s impressive.” Unlike the original Banik–Bertozzi ligands, arrayed as bulky chains on their polysaccharide backbones, Avilar’s bypass the need for multivalency to activate ASGPR. “We can dispense with the requirement of having a multimodal presentation,” says Grau. Smaller size enables oral delivery. (Lycia, says Staben, also now has monovalent receptor ligands.) One limitation: while ASGPR degraders can target any extracellular protein, they can only degrade membrane protein targets expressed on liver hepatocytes, where ASGPR is expressed.
But the mannose 6-phosphate receptor and ASGPR are just two effector molecules. The total number of internalizing receptors and other trafficking proteins may be vast. “[There] could end up being hundreds of proteins that one might be able to think about using as a way to get to lysosomes quickly,” says Banik, whose Stanford lab is working to identify potential protein shuttles.
Another outside-in approach for degrading membrane proteins recently emerged from Wells’s lab: AbTACs. AbTACS are bispecific antibodies that place a transmembrane E3 ligase next to a membrane protein of interest, sending it to the lysosome for degradation. (Some E3 ligases sort proteins for degradation in the lysosome instead of the proteasome.) Genentech’s PROTABs used the same membrane E3 ligases to efficiently degrade several cancer-driving membrane proteins in cells. The Genentech team also identified 38 different membrane E3 ligases, many of them cell type specific, that future degraders might use to shuttle membrane targets to lysosomes.
But AbTACs and PROTABs may not be able to degrade extracellular (secreted) proteins, and, like PROTACs, they have tricky ubiquitination requirements, so proximity alone may not induce degradation. Wells’s KineTACs, or cytokine-targeted chimeras, don’t have those limitations, degrading the extracellular protein VEGF (vascular endothelial growth factor) in addition to membrane proteins. All these degraders expand on LYTACs “by both providing additional degradation platforms and tissue selectivity,” Wells writes in an e-mail. Both AbTACs and KineTACs were licensed to EpiBiologics in October.
Can autophagy deliver?
For intracellular proteins, PROTACs and molecular glues — small-molecule E3 ligase binders that alter that enzyme’s surface to recruit a target protein for degradation — remain the dominant degrader modalities. But their inability to degrade aggregates, non-protein targets and organelles has inspired a set of degraders that instead hijack the autophagy system, which efficiently clears such debris from the cell.
Autophagy, which means “self-eating,” starts when the phagophore, a cup-shaped vesicle, scoops up cell contents. The phagophore closes around its cargo to form the double-membrane autophagosome vesicle, which then fuses with the lysosome and deposits its contents there for digestion. “These are normal pathways that we can just enhance further … with a small-molecule degrader,” says Casma’s Murphy.
Companies see potential for autophagy-based degraders in neurodegenerative diseases, particularly those characterized by mutant protein inclusions (Huntington’s), by intracellular tau aggregates (Alzheimer’s), or by α-synuclein fibrils and damaged mitochondria (Parkinson’s). Targeting mitochondria with a degrader in Parkinson’s or any condition is perilous, but conceivable. “Having a disease-specific marker on a mitochondrion [would] likely allow for a favorable therapeutic window,” says Murphy, who says several groups are looking for such markers. Non-neuronal protein aggregation disorders like amyloidosis are also in the picture. Lipid droplets could be targeted in metabolic disease. Even soluble proteins, which are not typically cleared by autophagy, are potential targets, says Murphy. “The majority of proteins in the cell, at some stage of their life, are associated with complexes,” he says. “The target space is broader than what you might think.”
The autophagy degrader space is so new that the effector molecules, analogous to the E3 ligases that PROTACs employ, are mostly undisclosed by the companies testing them. PAQ Therapeutics, for example, is developing ATTECs, or autophagosome-tethering compounds, using undisclosed autophagy proteins. Boxun Lu, a neuroscientist at Fudan University in Shanghai, found the first ATTECs when he conducted a screen for small-molecule binders to mutant huntingtin protein, which accumulates in the brain in Huntington’s disease, and counter-screened against LC3, a critical protein that recruits cargo to autophagosomes for transport to lysosomes. This screen improbably produced two molecular glues that bound to both. Lu’s group went on to show that these compounds and their more target-specific analogs could reduce mutant huntingtin and reverse disease in cells and in mouse disease models. Lu originally set out to find heterobifunctional molecules, not molecular glues, so “it is an extremely lucky event they were able to identify these molecules,” says Nan Ji, CEO of PAQ Therapeutics, which has licensed the technology.
These compounds are now in PAQ’s portfolio, but finding more single-warhead molecular glues for any given target usually involves a massive screening effort. So the company is developing ATTECs with dual linkers, one to an autophagy effector protein and the other a target of interest. These are simple to construct. But unlike the similarly two-headed PROTACs, which can employ known and validated binders, autophagy targets are mostly unexplored territory, and there are few if any known small-molecule binders to autophagy-related proteins.
Nan Ji says that PAQ has begun to break that barrier. Besides finding target binders, “we have also been able to identify very potent small-molecule binders to an autophagy protein,” he says. “I can’t really specify which one yet.”
Other companies are also keeping their autophagy proteins of choice under wraps. There are many possibilities, but also pitfalls, because autophagy is more complex than the ubiquitin–proteasome system, with a lot more players. In budding yeast there are at least 41 autophagy-related (Atg) proteins, which are highly conserved in mammals. A single degrader molecule that binds only one must recruit the entire autophagy machinery. Also, several different mechanisms of selective autophagy exist, suggesting that a single degrader platform may not be possible.
Murphy acknowledges the challenge. “It’s not a classical signaling pathway or cascade — it’s just not,” he says. “There are multiple components that sort of feed in at different levels; there’s a lot of cooperativity and parallel requirements.” Success, he says, will come from acquiring knowledge of specific autophagy factors sufficient to assemble the machinery for degradation. “I do believe it is going to be possible, once in possession of that knowledge, to deploy that approach across multiple therapeutic targets,” Murphy says. A universal autophagy degrader platform, he says, is possible. “This concept of self-sufficiency really holds the key.”
One autophagy degrader approach is the AUTAC, or autophagy-targeting chimera. Fifteen years ago, a group that included Hirokazu Arimoto, an organic chemist now at Tohoku University in Japan, discovered S-guanylation, a post-translational modification of proteins. Arimoto later observed that bacterial pathogens accumulate S-guanylated proteins around them and are cleared by autophagy, suggesting a mechanistic connection. To make the first AUTAC degraders, he linked an S-guanylation tag to a protein of interest, driving its degradation by attaching ubiquitin chains joined at their lysine 63 residues, a pattern that typically triggers autophagy. AUTACs degraded not only cytosolic proteins but also dysfunctional mitochondria, improving cellular energy production.
What’s missing from the AUTAC story is an explanation for how the S-guanylation post-translational modification triggers autophagy. “There’s a gap there,” says Murphy. “It’s unclear exactly how that molecule interfaces with the autophagy machinery.” It’s not known whether AUTACs have been licensed for commercial development; Arimoto did not respond to email messages from Nature Biotechnology.
Casma, like PAQ, has not revealed which autophagy protein or proteins it plans to use to effect target degradation. Autotac Bio, which is developing AUTOTACs, or autophagy-targeting chimeras, has disclosed one: p62, which forms a bridge between a protein that’s marked for degradation and the autophagy machinery. p62 and other autophagy receptors are not classical drug targets. “Finding appropriately tractable small molecule binding sites is going to be essential to success,” says Murphy. Using LC3 could be problematic because recent work has revealed non-autophagy functions for this family of proteins, so recruiting them to degrade any given target could impair some normal biology — for example, the transcription of genes for antimicrobial defense.
A risk exists that autophagy-based degraders could interfere with proteostasis by appropriating the autophagy machinery. “In a chronic disease setting, that’s something to really pay attention to,” says Murphy. “Hopefully it’s something we can modulate, because we’re using chemistry and pharmacology, and we can adjust dosing schedules appropriately to minimize that risk.”
Safety aside, will autophagy degrader drug discovery run into the same unpredictability problem as PROTACs? “There are too many unknowns to say we will definitely have an easier path here,” says Murphy. Nan Ji agrees. On the one hand, he says, even though an autophagy degrader, just like a PROTAC, must form a target–effector ternary complex, “there would be no need necessarily for ubiquitination to occur. So in theory, that this would save one requirement for degradation to happen.” On the other hand, autophagy degraders may sacrifice some specificity in the process. With PROTACs, “because of the requirement for ubiquitination, the lysine positioning, you could get additional selectivity — degradation selectivity,” Ji says. With ATACS, “that may not be the case.” Less built-in selectivity could mean more optimization steps for autophagy degraders, or lower potency. PAQ is still working out the discovery process.
Other TACs and TABs will emerge. “There are many paths to the lysosome,” says Banik. PROTACs are almost certain to find a niche in medical practice — molecular glues like the multiple myeloma drug Revlamid (lenalidomide) already have. But whether targeted degradation will become a major drug modality at the level of small-molecule inhibitors and antibodies is much less clear. Even less certain is the ultimate fate of LYTACs, ATACs, AbTACs, KineTACs, PROTABs, AUTACs, ATTECs and AUTOTACs. They’ve barely gotten started, and their biology is mostly unexplored. Companies, just out of stealth mode, are only now beginning to disclose their clinical candidates and preclinical results. The next year will be telling.
Ref:
