Source: https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/gene-expression-and-silencing/antisense-oligonucleotides
This article summarizes several of the common mechanisms of antisense gene modulation and more importantly, considerations to take into account when designing an antisense oligonucleotide (ASO). After decades of research, there are no hard and fast design rules; it is still trial and error. However, there are guidelines to be followed that should make the process more manageable.
Modulation Mechanisms
Traditional ASO-based gene modulation (usually synonymous with silencing or downregulation of gene expression, but it can be used to improve gene expression and, in at least one particular case, it was shown to lead to upregulation of gene expression) targets mRNA and can take place in either the nucleus or the cytoplasm. In the nucleus (pre-mRNA is the target), modulation typically works by redirecting polyadenylation, altering splicing events, or cleaving internucleotide bonds, all of which occur during mRNA maturation (Figure 1). In the cytoplasm (mature mRNA is the target), modulation typically works either by translational alteration without cleavage or cleavage, both of which occur just prior to / during translation (Figure 2).
ASO-based gene modulation mechanisms in the nucleus
Figure 1. ASO-based gene modulation mechanisms in the nucleus. In the case of mammals, gDNA in the nucleus is transcribed to pre-mRNA. An exogenous ASO in the nucleus hybridizes A) to the 3′-most polyadenylation signal on the pre-mRNA and blocks polyadenylation at this site, thereby redirecting it to another site upstream, which upregulates gene expression1 B) to a splice site, thereby preventing proper assembly of the spliceosome, which leads to exon skipping and therefore improved expression of a disease gene2 (not considered a true ASO by many, these are often called splice switching oligonucleotides [SSO] or more generally, steric blocking oligonucleotides [SBO]) C) an exon or intron (in this case, an intron), thereby leading to cleavage by RNase H3. In most cases, though significantly upregulated, silenced, or altered, some processing of the unaffected pre-mRNA is likely to occur followed by export of the mature mRNA to the cytoplasm. S = polyadenylation signal sequence (though only one is shown here, there can be more than one per transcript).
ASO-based gene modulation mechanisms in the cytoplasm
Figure 2. ASO-based gene modulation mechanisms in the cytoplasm.In the case of mammals, gDNA in the nucleus is 1) transcribed to pre-mRNA 2) pre-mRNA is processed (5′ cap and 3′ poly[A] tail are added) and spliced (introns are removed) to produce mature mRNA and 3) mature mRNA is exported to the cytoplasm. An exogenous ASO hybridizes to the mature mRNA in the cytoplasm and silences (downregulates) gene expression by A) translation alteration, in this case translation inhibition by disrupting ribosomal assembly at the 5′ cap4 (often not considered a true ASO, this is an example of a general SBO) or B) cleavage by RNase H (specifically, RNase H1 in humans)3. In most cases, though significantly downregulated, translation still occurs.
ASOs recognize and hybridize to target mRNAs by Watson-Crick base pairing. ASOs that lead to cleavage of target mRNAs by RNase H (whether in the nucleus or cytoplasm3) are widely studied for research and therapeutic purposes and therefore are the best understood in terms of modulation mechanism. Using magnesium ions as a cofactor, RNase H (specifically, RNase H1 in humans) cleaves the mRNA strand in the mRNA:DNA heteroduplex via hydrolysis of the internucleotide (phosphodiester) bond5. Following cleavage, the ASO remains intact while the former scissile bond is now free 3′-hydroxyl and 5′-phosphate groups on the 5′ and 3′ fragments, respectively, of the degraded mRNA.
Design Considerations
In principle, gene silencing should be as simple as selecting a sequence within a target mRNA; ordering the complementary, Watson-Crick-base-pairing ASO from a vendor; introducing it into the system under study (either in vitro or in vivo); and, observing the expected effect by the relevant reporter. However, there are many considerations to take into account for successful ASO design.
Hybridization Site
Following the rules of Watson-Crick base pairing, an ASO should hybridize to any region of a target mRNA sequence. However, mRNAs fold into secondary and even tertiary structures, which likely block ASO hybridization. Therefore, non-folded regions of mRNAs should be selected as the hybridization sites. There are wet-laboratory methods, such RNase H mapping that are useful in predicting an accessible site, but a good place to start is to try a predictive RNA folding algorithm, e.g. mfold.
Once a non-folded region has been identified, a secondary consideration should be if the region serves as a binding site for spliceosomes, ribosomes, proteins, or other macromolecular assemblies. Historically, the 5′ cap, initiation codon, 3′ untranslated region / polyA tail have been good site selections. Even if the ASO fails to activate RNase H, it may still lead to silencing since it will sterically block the machinery needed for mRNA maturation or translation.
Nuclease Degradation
In vivo and in vitro, all-native DNA ASOs are quickly rendered useless by nuclease activity. In vivo, though both endonucleases and exonucleases may lead to degradation, exonucleases appear to do most of the damage. To be effective, all ASOs require chemical modification to resist nuclease degradation. Though numerous nucleic acid analogs are available for modifying ASOs, herein only those that are part of our standard modifications offering will be explored (Table 1).
Three regions of ASOs are subject to modification (internucleotide linkages, sugars, and bases), and in all subsequent sections, modifications are classified according to their primary effect, even though several have more than one effect, e.g. modification X primary effect: improves binding affinity; secondary effect: reduces the deleterious impact from immunostimulation (the focus of this article will remain on the primary effect).
Modification by Type
Internucleotide Linkages
Chemistry Abbreviation Structure
Phosphorothioate (aka Thiophosphate or S-oligo) PS (* in sequence constructs)
Sugars
Chemistry Abbreviation Structure
Methyl RNA 2′-OMe-RNA ([mA], [mC], [mG], & [mU] in sequence constructs)
mA
Table 1Available modifications that are primarily intended to resist nuclease degradation.
Phosphorothioate. This modification was among the few that is considered first-generation. PS-ASOs are nuclease resistant and, therefore, have longer plasma half lives compared to all-native DNA ASOs. In addition, they retain negative backbone charges, which facilitates PS-ASO entry into the cell. Interestingly, PS appears to have a bigger impact on transport and entry into the cell than it does on nuclease resistance.
However, PS-ASOs are not completely protected from nucleases, have reduced hybridization to target mRNAs (see the Binding Affinity section), and must be continually administered in large quantities to maintain modulation. In addition, PS can interact with proteins in vivo and therefore lead to negative side effects, including immune system activity.
Methyl RNA. This modification was among the few that are considered second-generation. When combined with PS in ASOs, 2′-OMe-RNA has been found to improve upon the benefits of PS alone (i.e. increased nuclease resistance, plasma half life, and tissue uptake).
Immunostimulation
Bacterial DNA contains a much higher frequency of CpG (cytosine-phosphodiester bond-guanine) dinucleotides lacking methylation than does vertebrate DNA. This is primarily because CpG dinucleotides are underrepresented in the vertebrate genome and 80% of them are labeled with methyl groups. Since the CpG motif in bacteria triggers activation of B cells, NK cells, monocytes, and cytokines whereas the vertebrate CpG motif does not, this is likely at least one of the ways that the immune system recognizes a bacterial infection. ASOs containing unmethylated CpG (CpsG: cytosine-phosphorothioate bond-guanine is even more potent) motifs stimulate the immune system in a manner similar to that of bacterial DNA and may have been responsible for some reported effects from early antisense studies.
To avoid immunostimulation, design ASOs lacking CpG / CpsG motifs, if possible, or least those lacking the following extended motif, which produces the strongest immune response:
purine-purine-CpG-pyrimidine-pyridmidine
Given that this may be difficult to avoid due to the complementary nature of the target site selection sequence, the next best step is to replace the cytosine in CpG / CpsG with 5’-methylcytosine (Table 2), which has been shown to decrease immunostimulation significantly.
Modification by Type
Bases
Chemistry Abbreviation Structure
5-methylcytosine 5-Me-dC ([5MedC] in sequence constructs)
Table 2Available modification that is primarily intended to prevent immunostimulation.
Sequence Length
Optimum lengths are usually from 12 to 28 bases. Sequences shorter than 12 bases increase the probability of off-target hybridization, while sequences longer than 25 bases increase the chance of reduced cellular uptake.
Self Complementarity
The ASO should be checked for secondary structure and oligonucleotide dimer formation as either one might interfere with hybridization to the target site sequence. If possible, design the ASO to have the weakest secondary structure possible as well as no dimer formation. Our oligonucleotide sequence calculator OligoEvaluator™ allows for quick determination of these self-forming structures.
G-Quartet Structures
ASOs containing stretches of two or more C or G nucleotides are able to form unusual structures, which may produce undesirable, off-target effects. The most common and studied are stretches of G bases, which can lead to the formation of G-quartets. These quartets have been shown to bind to proteins, including transcription factors, which may mimic and therefore interfere with antisense activity.
To avoid formation of these quartets, design ASOs lacking these polyG stretches, if possible. Again, given that this may not be feasible, the next best step is to replace the guanine with 7-deaza-dG (Table 3), which will block quartet formation.
Modification by Type
Bases
Chemistry Abbreviation Structure
7-deaza-dG [Deaza-dG] in sequence constructs (not available for online ordering, so please inquire)
Table 3Available modification that is primarily intended to prevent G-quartet formation.
Functional Motifs
A statistical analysis of PS-ASO experiments found that the following motifs:
correlate with enhanced antisense efficiency, whereas these motifs:
diminish antisense activity. It has been found that RNase H activity is sequence independent; Therefore, it is believed that the enhancing motifs lead to increased thermodynamic stability of the mRNA:ASO heteroduplex through the preponderance of GC Watson-Crick base pairing.
Binding Affinity
As already discussed, it is critically important to identify a site within the target mRNA that is free of folds as well as to ensure that the ASO also has no deleterious self complementarity. However, these considerations alone are not enough to ensure proper hybridization. Various factors, such as PS can reduce ASO binding affinity for the target site, which in turn minimizes antisense effectiveness.
Third-generation ASO modifications have been found not only to be nuclease resistant but also to improve binding affinity. Locked Nucleic Acid® (Table 4), with its constrained ring structure, is particularly useful for improving ASO binding affinity and effectiveness (melting temperature change per monomer addition varies from +3 to +11 °C compared to native DNA only).
Modification by Type
Bases
Chemistry
Locked Nucleic Acid
([+A], [+C], [+G], & [+T] in sequence constructs; not currently available for online ordering, so please inquire)
Table 4Available modification that is primarily intended to improve ASO binding affinity.
The Construct
To give insight into ASO sequences, examples of several antisense drugs (often the primary purpose of pursuing antisense research) that have been approved or are in clinical trials are provided here. These drugs are examples of (or are expected to be in the case of those in clinical trials) all of the desired outcomes when it comes to antisense: good design, an available delivery mechanism, and effective modulation. The same outcomes are critical to the success of research experiments (our ASOs are for in vitro and in vivo animal RUO [Research Use Only]).
First generation. In 1998, Fomivirsen (brand name Vitravene) was the first approved antisense drug. It was used to treat cytomegalovirus retinitis (CMV) in immunocompromised patients, including those with AIDS. The drug was delivered by intravitreal injection. The 21mer ASO with all PS internucleotide linkages has the following sequence:
G*C*G*T*T*T*G*C*T*C*T*T*C*T*T*C*T*T*G*C*G
○ * = PS
and works by inhibiting translation of transcribed mRNA from the CMV gene UL123. It was eventually withdrawn from the market because the development of HAART (highly active antiretroviral therapy) to treat HIV reduced the number of CMV cases by 75% and therefore led to poor sales.
Since PS-only ASOs are not completely protected from nucleases, have reduced hybridization to target mRNAs, must be continually administered in large quantities to maintain modulation, and can interact with proteins, which may lead to negative side effects, first-generation constructs have largely been abandoned in R&D pipelines.
Second generation. In 2013, Mipomersen (brand name Kynamro®) became the second approved antisense drug. It is used to treat familial hypercholesterolemia, a hereditary disorder. The drug is delivered by subcutaneous injection. The 20mer ASO with all PS internucleotide linkages has the following sequence:
G*mC*mC*mU*mC*A*G*T*mC*T*G*mC*T*T*mC*G*mC*A*mC*mC
○ Underline = 2′-O-MOE-RNA (MOE is 2-methoxyethyl)
○ m = methyl, i.e. 5-Me-dC & 5-Me-U
○ * = PS
and works by inhibiting translation of apolipoprotein B-100 mRNA23. There is a risk of severe liver damage, so the drug has to be part of a risk management plan.
Second-generation antisense molecules, such as Mipomersen, are designed with the 5-10-5 gapmer configuration. This can be seen in the sequence above: 5′ and 3′ wings of 5 bases (modified with a nuclease-resistant / enhanced-binding-affinity sugar modification) and a central gap of 10 standard deoxyribonucleotides (no sugar modification) that allows for RNase H binding.
In this particular case, the wings consist of 2′-O-MOE-RNA (MOE is 2-methoxyethyl), a non-standard sugar modification. However, we might be able to add this to your construct, so please send a request to dnaoligos@sial.com for feasibility.
Third generation. As of 2017, Miravirsen (SPC3649) is in Phase II clinical trials. It is being tested as a treatment for hepatitis C (HCV). The drug is delivered by subcutaneous injection. The 15mer ASO with all PS internucleotide linkages has the following sequence:
C*C*A*T*T*G*T*C*A*C*A*C*T*C*C
○ Underline = LNA
○ * = PS
and works by hybridizing to human miRNA, miR-122. This prevents miR-122 from bringing argonaute to the 5′-UTR region of the HCV RNA, where it normally binds and therefore protects against nuclease degradation. Therefore, Miravirsen allows for destruction of the viral RNA.
Though Miravirsen is not a traditional ASO as it targets miRNA and therefore only indirectly leads to degradation of mRNA, it is one of the best examples of a third-generation construct containing LNA, hence it is included here.
Target Check
The final non-modified ASO sequence should be put through a BLAST search to ensure that any off-target hybridization — preferrably none — will not interfere with antisense activity or lead to unacceptable toxicity.
Quality Considerations
For in vivo animal experiments, we recommend ASOs undergo in-vivo-grade purification with a salt exchange (replaces toxic ammonium ions from the phosphoramidite synthesis chemistry with physiological sodium ions), endotoxin testing (ensures that pyrogens are present below an acceptable ceiling), and filtration (reduces the number of contaminating CFU below an acceptable ceiling). Our iScale Oligos™ product is larger quantities of material for in vivo projects that can be ordered with this purification and all of these additional services.
Delivery & Toxicity
Though beyond the scope of this article, there are several excellent review papers that discuss various delivery mechanisms as well as potential toxicities.
Conclusion
When you have designed an ASO that you want to try in an experiment, we are ready to synthesize it for you (our ASOs are for in vitro and in vivo animal RUO [Research Use Only]). If additional help is needed, especially regarding the feasibility of manufacturing ASOs with non-standard modifications, please send a request to dnaoligos@sial.com.
References
1.
Vickers TA. 2001. Fully modified 2′ MOE oligonucleotides redirect polyadenylation. 29(6):1293-1299. http://dx.doi.org/10.1093/nar/29.6.1293
2.
Kole R, Krainer AR, Altman S. 2012. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 11(2):125-140. http://dx.doi.org/10.1038/nrd3625
3.
Liang X, Sun H, Nichols JG, Crooke ST. 2017. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Molecular Therapy. 25(9):2075-2092. http://dx.doi.org/10.1016/j.ymthe.2017.06.002
4.
Baker BF, Lot SS, Condon TP, Cheng-Flournoy S, Lesnik EA, Sasmor HM, Bennett CF. 1997. 2?-O-(2-Methoxy)ethyl-modified Anti-intercellular Adhesion Molecule 1 (ICAM-1) Oligonucleotides Selectively Increase the ICAM-1 mRNA Level and Inhibit Formation of the ICAM-1 Translation Initiation Complex in Human Umbilical Vein Endothelial Cells. J. Biol. Chem.. 272(18):11994-12000. http://dx.doi.org/10.1074/jbc.272.18.11994
5.
Deleavey G, Damha M. 2012. Designing Chemically Modified Oligonucleotides for Targeted Gene Silencing. Chemistry & Biology. 19(8):937-954. http://dx.doi.org/10.1016/j.chembiol.2012.07.011
6.
Patil SD, Rhodes DG, Burgess DJ. 2005. DNA-based therapeutics and DNA delivery systems: A comprehensive review. AAPS J. 7(1):E61-E77. http://dx.doi.org/10.1208/aapsj070109
7.
Goodchild J, Carroll E, Greenberg JR. 1988. Inhibition of rabbit ?-Globin synthesis by complementary oligonucleotides: Identification of mRNA sites sensitive to inhibition. Archives of Biochemistry and Biophysics. 263(2):401-409. http://dx.doi.org/10.1016/0003-9861(88)90652-2
8.
Fisher TL, Terhorst T, Cao X, Wagner RW. 1993. Intracellular disposition and metabolism of fluorescently-labled unmodified and modified oligouncleotides microijjected into mammalian cells. Nucl Acids Res. 21(16):3857-3865. http://dx.doi.org/10.1093/nar/21.16.3857
9.
EDER PS, DeVINE RJ, DAGLE JM, WALDER JA. 1991. Substrate Specificity and Kinetics of Degradation of Antisense Oligonucleotides by a 3? Exonuclease in Plasma. Antisense Research and Development. 1(2):141-151. http://dx.doi.org/10.1089/ard.1991.1.141
10.
DAGLE JM, WEEKS DL, WALDER JA. 1991. Pathways of Degradation and Mechanism of Action of Antisense Oligonucleotides inXenopus laevisEmbryos. Antisense Research and Development. 1(1):11-20. http://dx.doi.org/10.1089/ard.1991.1.11
11.
Mansoor M, Melendez AJ. 2008. Advances in Antisense Oligonucleotide Development for Target Identification, Validation, and as Novel Therapeutics. Gene?Regul Syst Bio. 2GRSB.S418. http://dx.doi.org/10.4137/grsb.s418
12.
Khvorova A, Watts JK. 2017. The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol. 35(3):238-248. http://dx.doi.org/10.1038/nbt.3765
13.
Weiner GJ, Liu H, Wooldridge JE, Dahle CE, Krieg AM. 1997. Immunostimulatory oligodeoxynucleotides containing the CpG motif are effective as immune adjuvants in tumor antigen immunization. Proceedings of the National Academy of Sciences. 94(20):10833-10837. http://dx.doi.org/10.1073/pnas.94.20.10833
14.
KRIEG AM, MATSON S, FISHER E. 1996. Oligodeoxynucleotide Modifications Determine the Magnitude of B Cell Stimulation by CpG Motifs. Antisense and Nucleic Acid Drug Development. 6(2):133-139. http://dx.doi.org/10.1089/oli.1.1996.6.133
15.
Henry S, Stecker K, Brooks D, Monteith D, Conklin B, Bennett C. 2000. Chemically modified oligonucleotides exhibit decreased immune stimulation in mice.. J Pharmacol Exp Ther. 292468-79. https://pubmed.ncbi.nlm.nih.gov/10640282/
16.
Williamson JR, Raghuraman M, Cech TR. 1989. Monovalent cation-induced structure of telomeric DNA: The G-quartet model. Cell. 59(5):871-880. http://dx.doi.org/10.1016/0092-8674(89)90610-7
17.
Tam R, Lin C, Lim C, Pai B, Stoisavljevic V. 1999. Inhibition of CD28 expression by oligonucleotide decoys to the regulatory element in exon 1 of the CD28 gene.. J Immunol. 1634292-9. https://pubmed.ncbi.nlm.nih.gov/10510368/
18.
Murchie A, Lilley D. 1994. Tetraplex folding of telomere sequences and the inclusion of adenine bases.. The EMBO Journal. 13(4):993-1001. http://dx.doi.org/10.1002/j.1460-2075.1994.tb06344.x
19.
Matveeva OV. 2000. Identification of sequence motifs in oligonucleotides whose presence is correlated with antisense activity. 28(15):2862-2865. http://dx.doi.org/10.1093/nar/28.15.2862
20.
Ho S. 1996. Potent antisense oligonucleotides to the human multidrug resistance-1 mRNA are rationally selected by mapping RNA-accessible sites with oligonucleotide libraries. 24(10):1901-1907. http://dx.doi.org/10.1093/nar/24.10.1901
21.
Wahlestedt C, Salmi P, Good L, Kela J, Johnsson T, Hokfelt T, Broberger C, Porreca F, Lai J, Ren K, et al. 2000. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proceedings of the National Academy of Sciences. 97(10):5633-5638. http://dx.doi.org/10.1073/pnas.97.10.5633
22.
Mulamba GB, Hu A, Azad RF, Anderson KP, Coen DM. 1998. Human Cytomegalovirus Mutant with Sequence-Dependent Resistance to the Phosphorothioate Oligonucleotide Fomivirsen (ISIS 2922). Antimicrob. Agents Chemother.. 42(4):971-973. http://dx.doi.org/10.1128/aac.42.4.971
23.
Geary RS, Baker BF, Crooke ST. 2015. Clinical and Preclinical Pharmacokinetics and Pharmacodynamics of Mipomersen (Kynamro®): A Second-Generation Antisense Oligonucleotide Inhibitor of Apolipoprotein B. Clin Pharmacokinet. 54(2):133-146. http://dx.doi.org/10.1007/s40262-014-0224-4
24.
Liang X, Sun H, Nichols JG, Crooke ST. 2017. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Molecular Therapy. 25(9):2075-2092. http://dx.doi.org/10.1016/j.ymthe.2017.06.002
25.
Titze-de-Almeida R, David C, Titze-de-Almeida SS. 2017. The Race of 10 Synthetic RNAi-Based Drugs to the Pharmaceutical Market. Pharm Res. 34(7):1339-1363. http://dx.doi.org/10.1007/s11095-017-2134-2
26.
Gebert LFR, Rebhan MAE, Crivelli SEM, Denzler R, Stoffel M, Hall J. 2014. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. 42(1):609-621. http://dx.doi.org/10.1093/nar/gkt852
27.
Chan JH, Lim S, Wong WF. 2006. ANTISENSE OLIGONUCLEOTIDES: FROM DESIGN TO THERAPEUTIC APPLICATION. Clin Exp Pharmacol Physiol. 33(5-6):533-540. http://dx.doi.org/10.1111/j.1440-1681.2006.04403.x
28.
Godfrey C, Desviat LR, Smedsrød B, Piétri?Rouxel F, Denti MA, Disterer P, Lorain S, Nogales?Gadea G, Sardone V, Anwar R, et al. 2017. Delivery is key: lessons learnt from developing splice?switching antisense therapies. EMBO Mol Med. 9(5):545-557. http://dx.doi.org/10.15252/emmm.201607199
29.
Sun Y, Zhao Y, Zhao X, Lee RJ, Teng L, Zhou C. Enhancing the Therapeutic Delivery of Oligonucleotides by Chemical Modification and Nanoparticle Encapsulation. Molecules. 22(10):1724. http://dx.doi.org/10.3390/molecules22101724
30.
Krhac Levacic A, Morys S, Wagner E. 2017. Solid-phase supported design of carriers for therapeutic nucleic acid delivery. 37(5): http://dx.doi.org/10.1042/bsr20160617

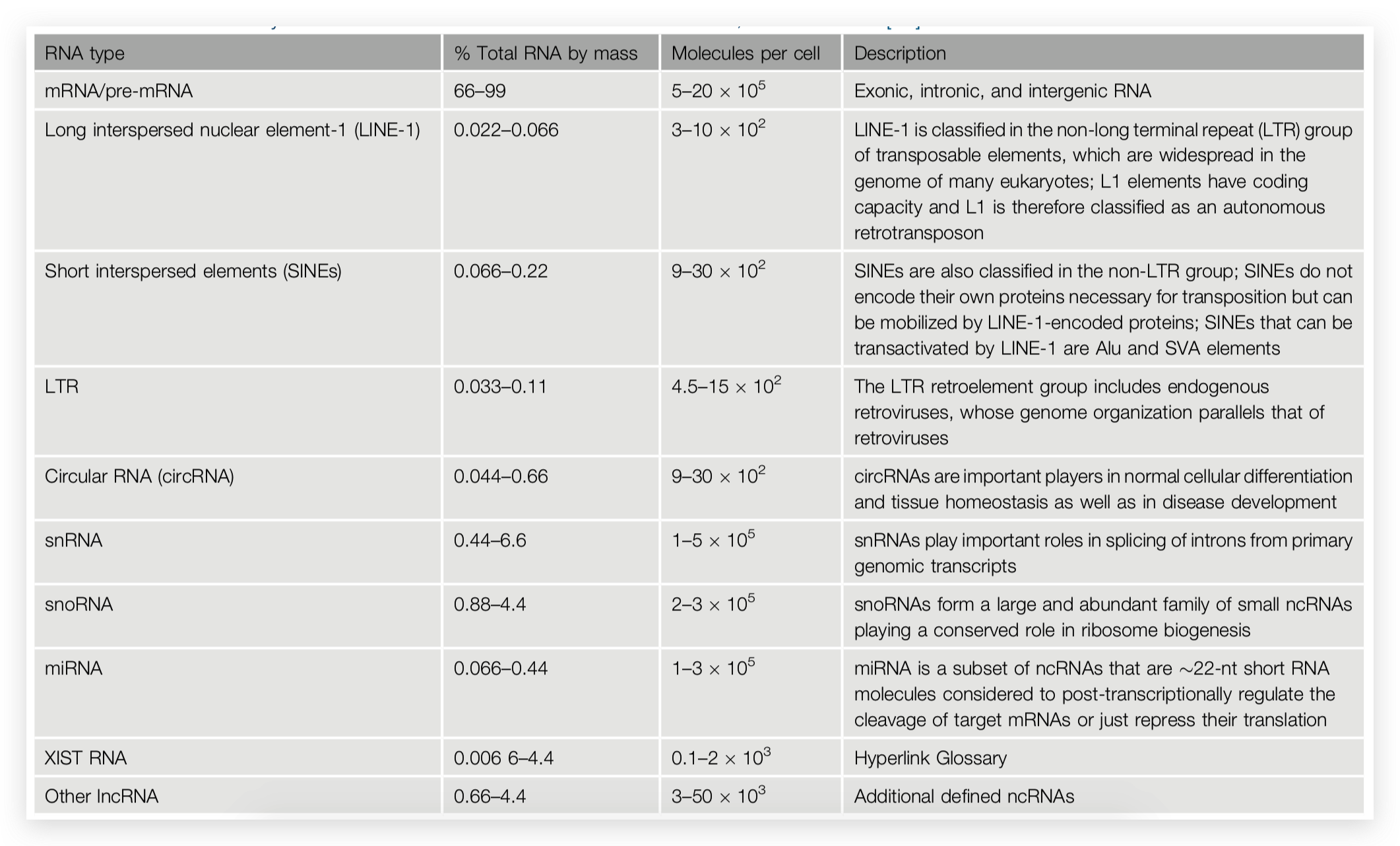
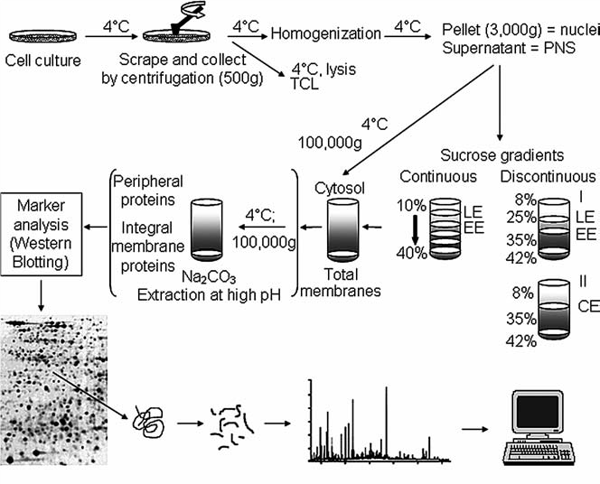 Figure 1: Schematic outline of fractionation and isolation of endosomal organelles using gradient based ultracentrifugation followed by proteome analysis. Source: Araújo et al., Methods in Molecular Biology. 2008.
Figure 1: Schematic outline of fractionation and isolation of endosomal organelles using gradient based ultracentrifugation followed by proteome analysis. Source: Araújo et al., Methods in Molecular Biology. 2008.
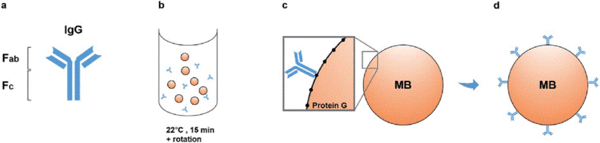
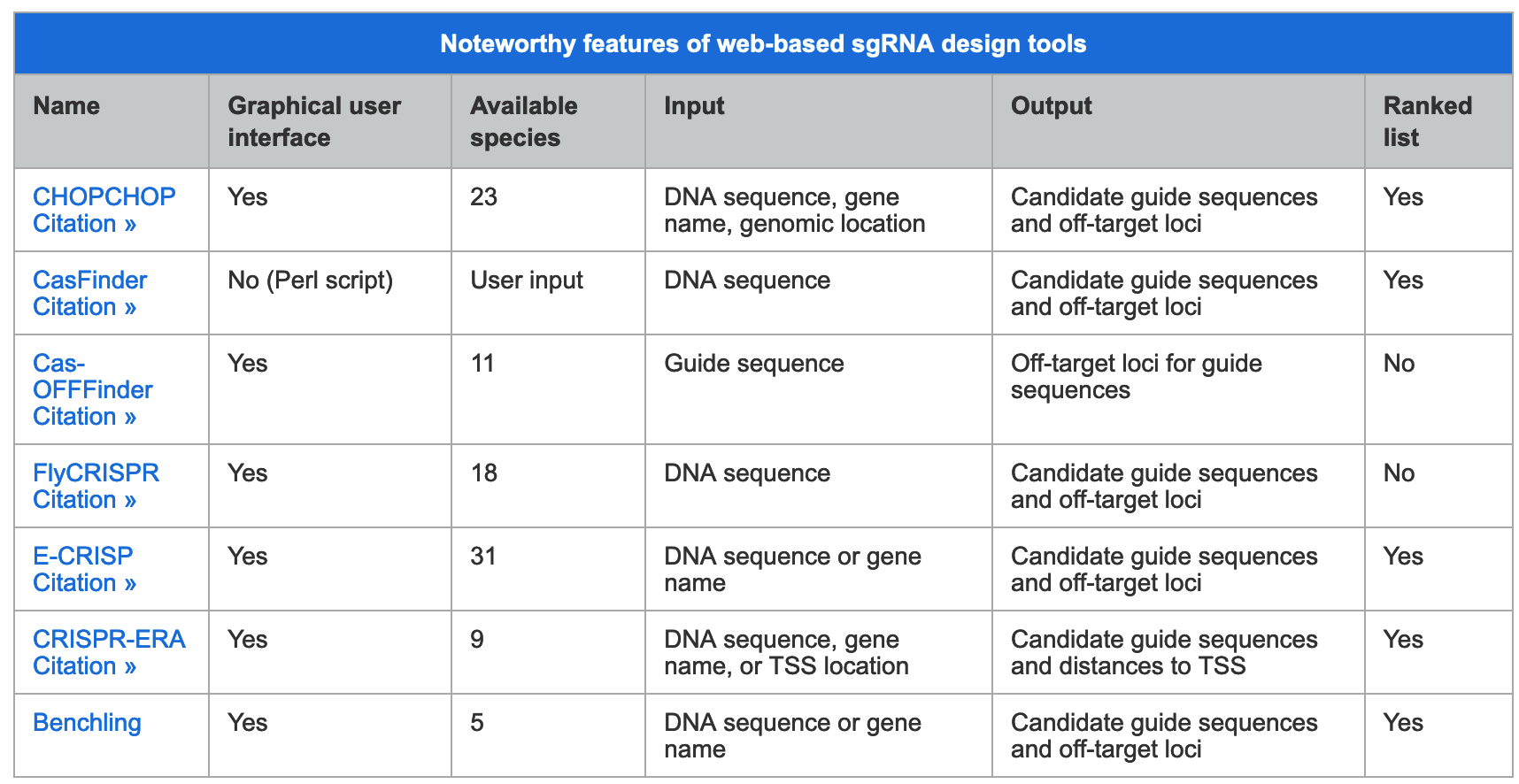

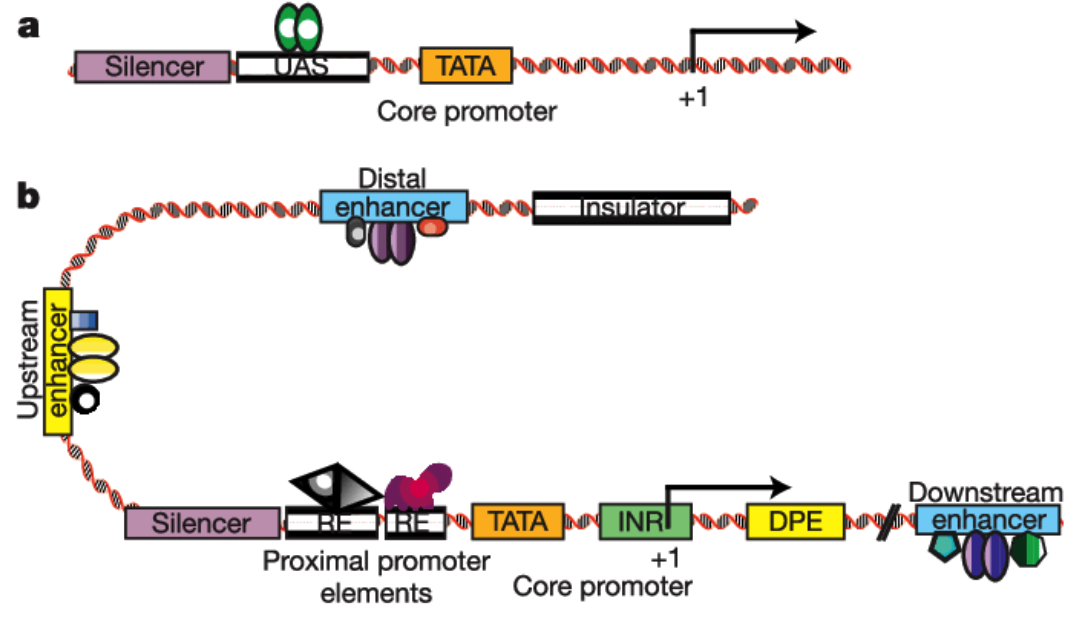
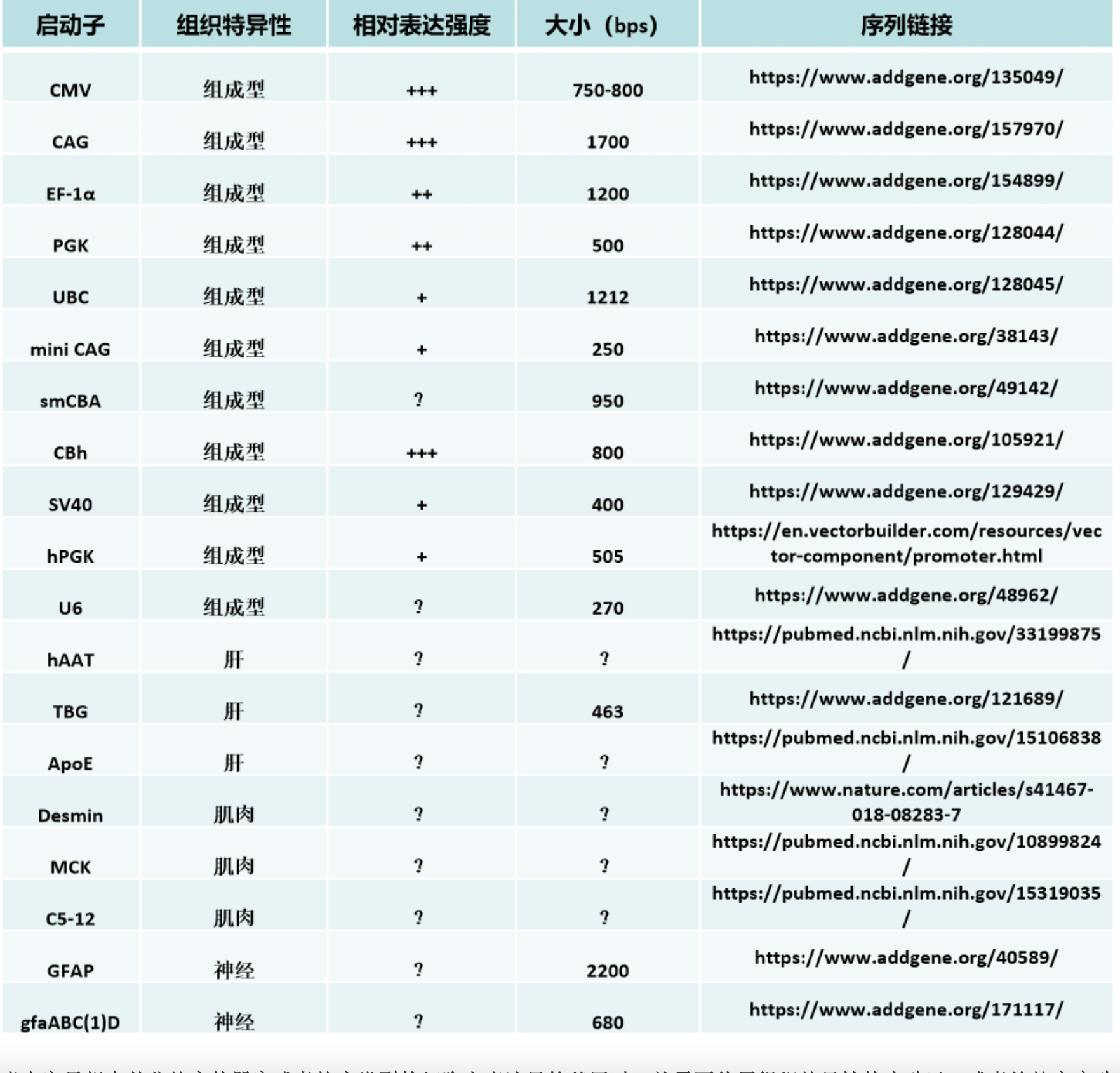




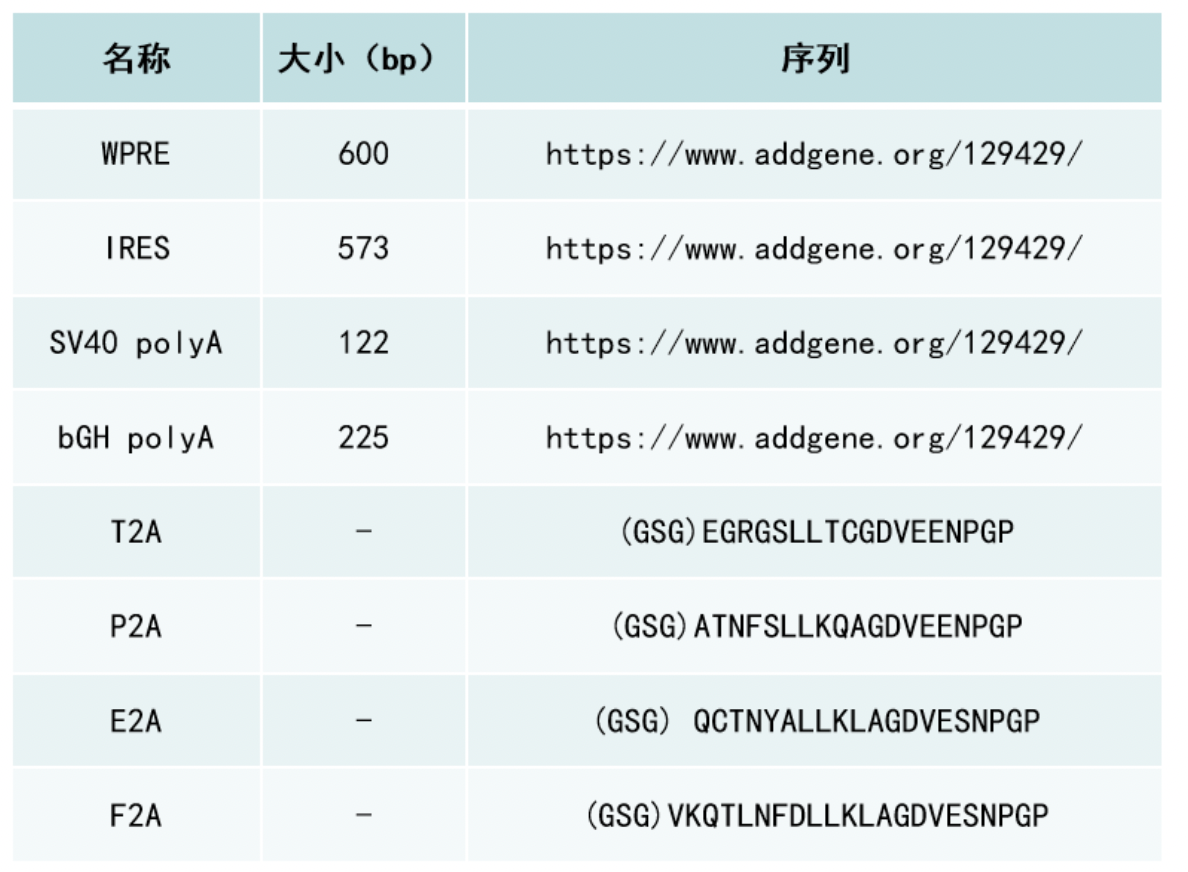

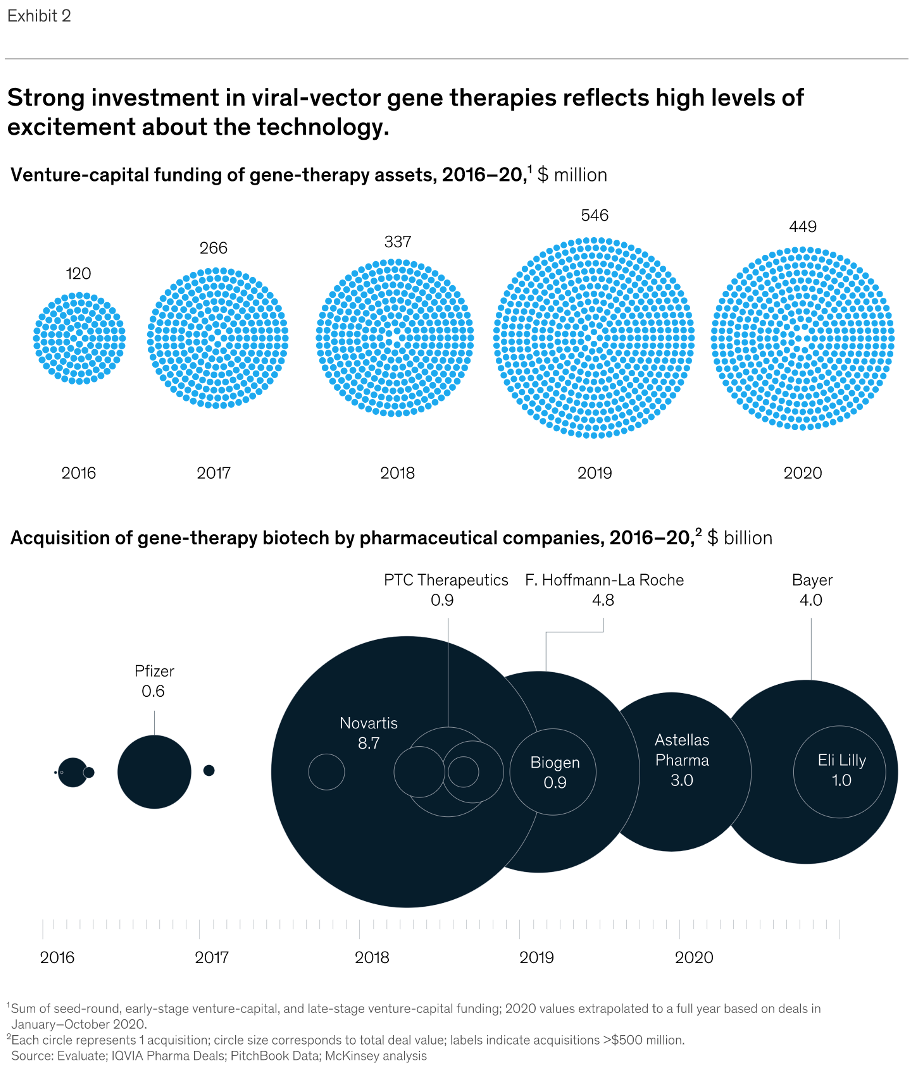



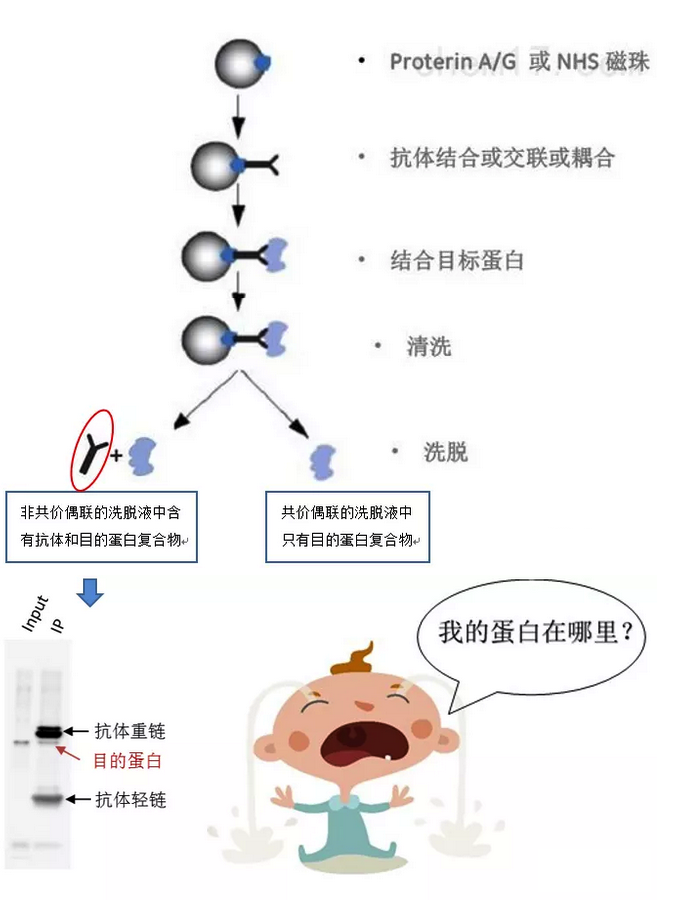
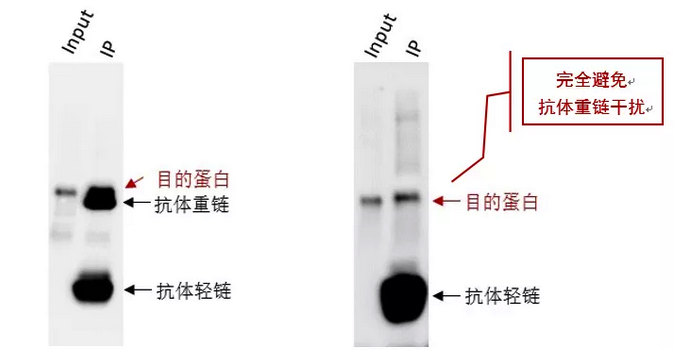


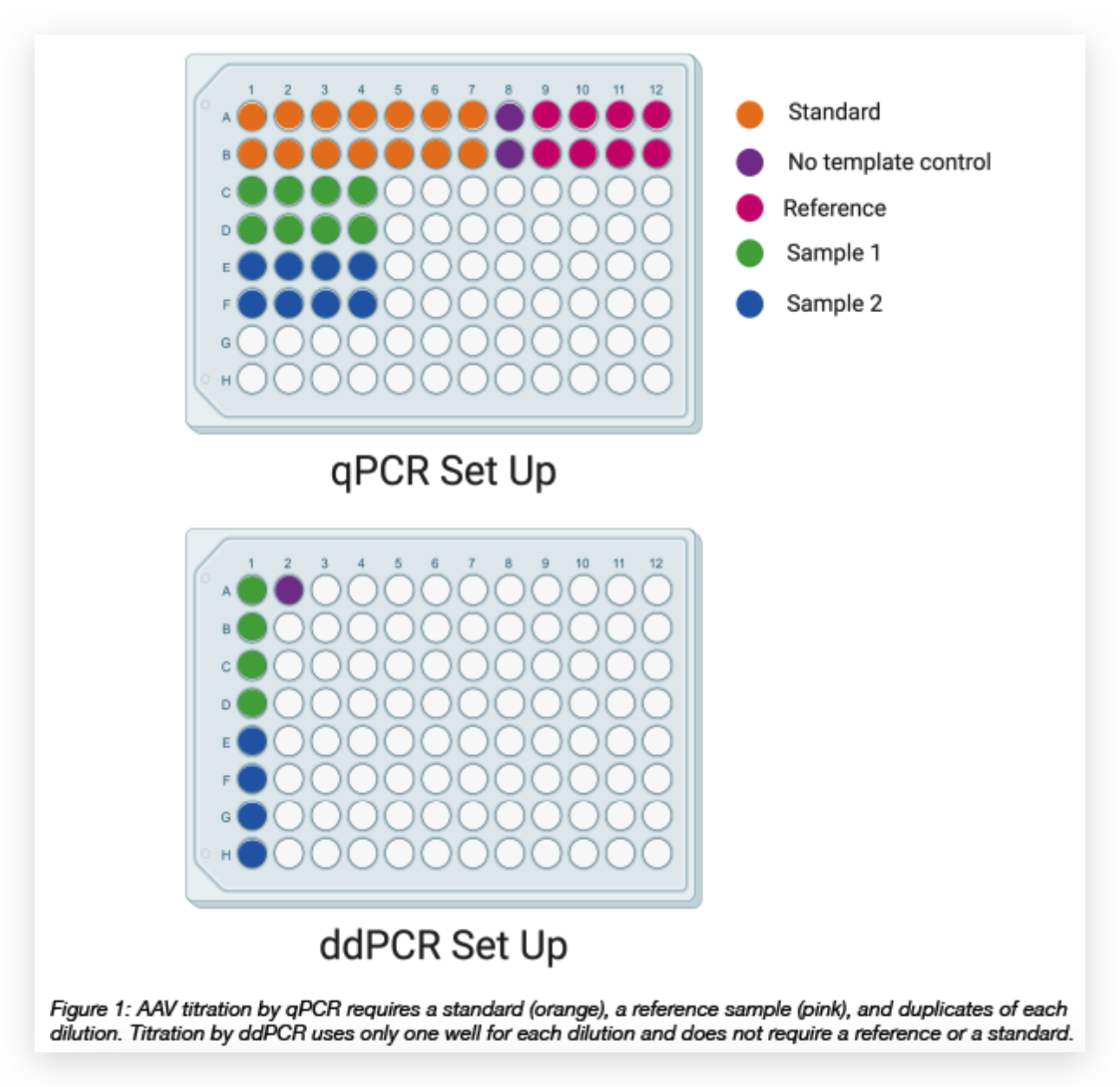 We have also found that generating the standard curve for qPCR can be challenging to get right. We use a linearized plasmid standard. However, even when the standard is made properly, it is unstable and may only work for a small number of qPCR runs before a new one is needed. For these reasons, we are in the process of transitioning our titering method from qPCR to ddPCR.
We have also found that generating the standard curve for qPCR can be challenging to get right. We use a linearized plasmid standard. However, even when the standard is made properly, it is unstable and may only work for a small number of qPCR runs before a new one is needed. For these reasons, we are in the process of transitioning our titering method from qPCR to ddPCR.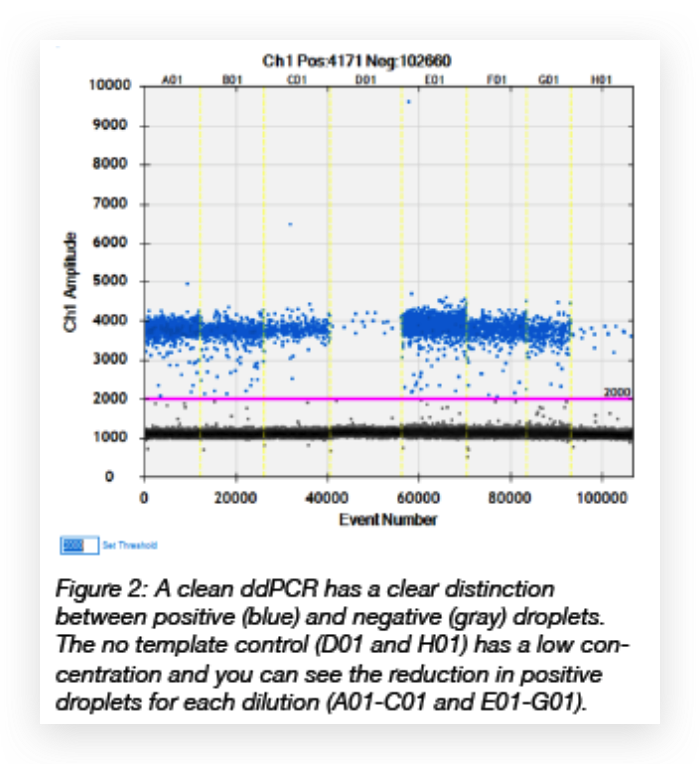 The ddPCR software uses the ratio of positive to negative droplets is used to calculate the concentration of the sample. This concentration can then be used to calculate the viral titer:
The ddPCR software uses the ratio of positive to negative droplets is used to calculate the concentration of the sample. This concentration can then be used to calculate the viral titer:


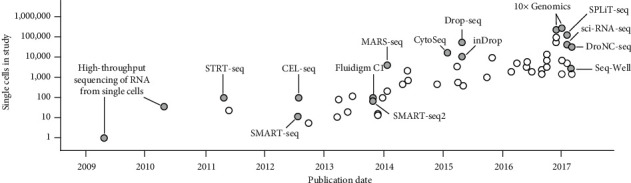
 RNA-seq is based on next-generation sequencing (NGS) and allows for discovery, quantitation and profiling of RNA. The
RNA-seq is based on next-generation sequencing (NGS) and allows for discovery, quantitation and profiling of RNA. The